【7.3.5】使用 BLAST 验证质粒的技巧
选择 BLAST 程序
在可用的五个 BLAST 程序中,我们主要使用标准核苷酸 BLAST (blastn)、标准蛋白质 BLAST (blastp) 和 Translated BLAST (blastx)。NCBI 有一个很棒的 BLAST 入门指南,其中包括对不同 BLAST 程序、数据库和 BLAST 搜索页面元素的简单解释。
在 Addgene,我们使用blastn 来识别Sanger 序列中的任何差异,例如错配、缺失或插入。我们使用blastp 或blastx 将我们的测序结果与蛋白质序列进行比较,以检查开放阅读框(ORF)并确定任何核苷酸差异的潜在影响。blastp 和blastx 程序的优化方式不同,您可能希望根据要验证的信息选择一个(或两个)。我们将在下面深入探讨这些差异。
优化Blastn搜索
在标准核苷酸 BLAST页面上,首先要做的决定是将 Sanger 测序结果与单个已知参考序列还是与 BLAST 序列数据库进行比较。如果您知道预期的核苷酸序列,请选中“对齐两个或多个序列”复选框并将您的参考序列粘贴到出现的主题序列框中。比对两个核苷酸序列可能是执行速度最快的 BLAST 搜索,与其他类型的 BLAST 搜索相比,可以节省您的时间。
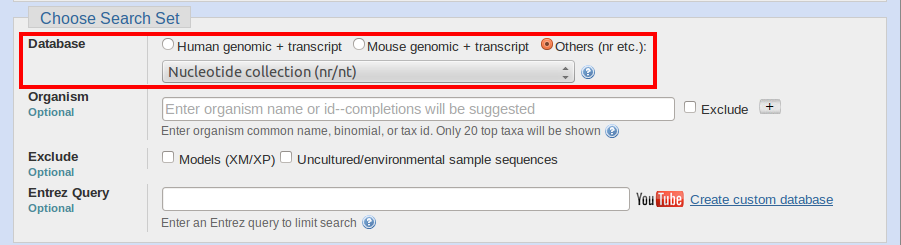
如果您不知道结果的确切参考序列,请从下拉菜单中选择 BLAST 序列数据库之一。通常,我们使用默认的核苷酸数据库“核苷酸集合 (nr/nt)”,因为它包含 GenBank、EMBL、DDBJ 和 PDB 序列的组合,并且可能是最全面的搜索。
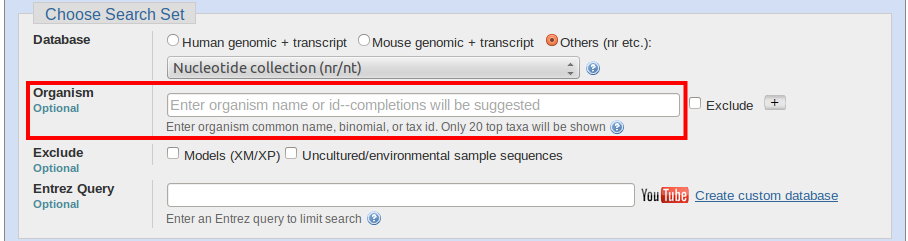
省时技巧 #1:如果您知道测序结果应匹配的物种,请在有机体框中输入通用名称或科学名称。这条小信息可以显着减少您对blastn、blastp 和blastx 搜索的等待时间!
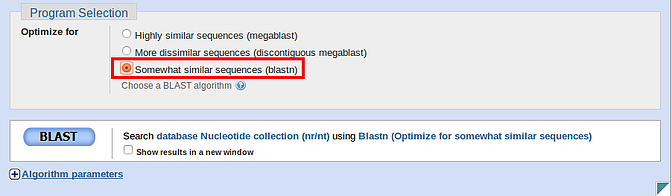
相似的序列(megablast)”,当您的序列与参考/数据库序列之间的同一性≥ 95% 时,该设置非常快且效果最佳。[如果我们 95% 的结果总是正确的,我们的 QC 过程将是无故障且更快的!]
由于 Sanger 测序反应不完美,并且反应开始或结束附近的序列通常不可靠,我们建议对 Blastn 使用“有点相似的序列 (blastn)”程序,以便您可以从结果中提取几乎每一个可靠的碱基对。
此选项不如 megablast 快,但可以返回更长的比对以与您的测序跟踪文件进行比较。与 megablast 不同的是,常规的 Blastn 程序使用更小的字长和更低的比对中的不匹配和差距的得分惩罚。如果您对blastn 程序的差异感到好奇,请查看BLAST 帮助网页。
优化blastx搜索 Optimizing blastx searches
使用blastn确定Sanger 测序结果的可靠部分并注意到核苷酸序列中的任何潜在错配或缺口后,您可以运行 Translated BLAST (blastx) 搜索以检查预期的ORF、突变或截断。blastx 的一个主要优点是您不必为测序结果决定阅读框——blastx 根据数据库检查所有六个可能的框。另一个好处是,当查看blastx 结果时,ORF 中存在的移码突变很明显。
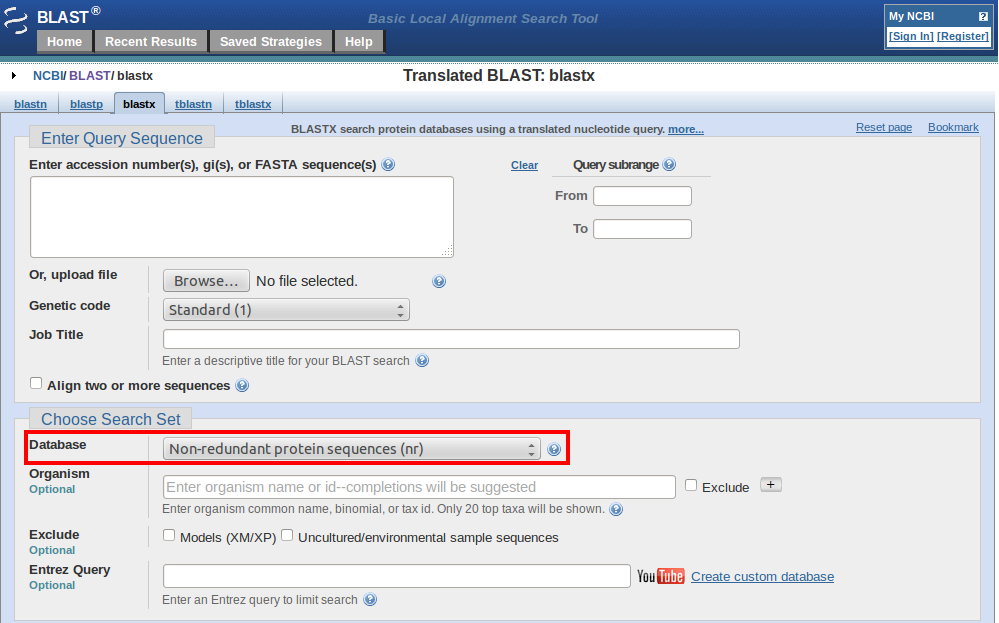
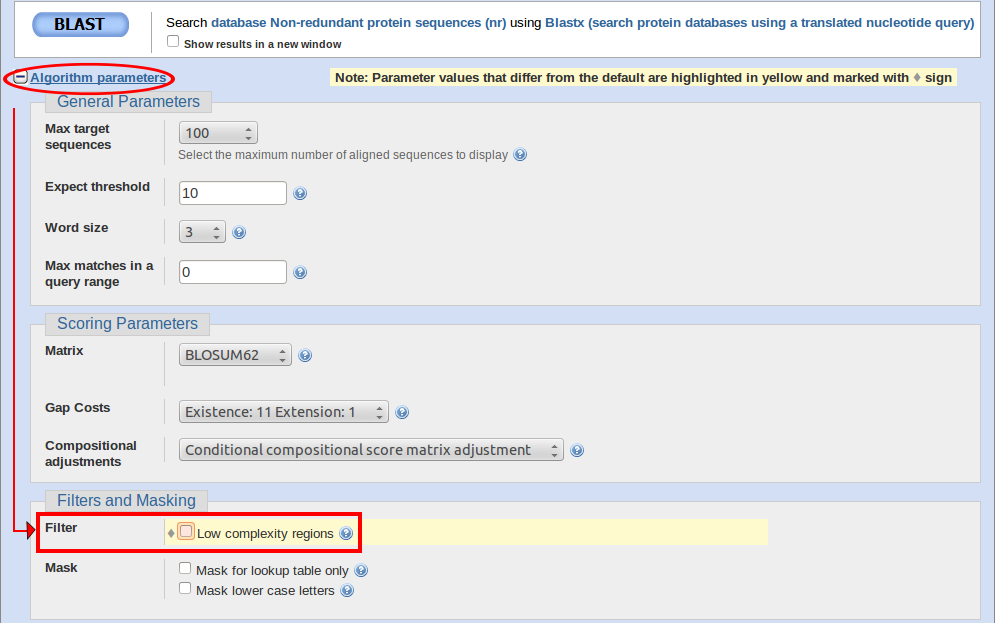
使用blastx时在 Addgene,我们使用默认的“非冗余蛋白质序列 (nr)”数据库,因为它包含最多数量的蛋白质序列。在 BLAST 按钮下方,您可能已经注意到“算法参数”链接。单击此链接可查看高级 BLAST 选项以及我们建议的blastx 自定义。与核苷酸序列类似,蛋白质通常具有重复或高度同源的区域,在标准的blastx 搜索中默认会忽略这些区域。省略重复区域的比对可能会令人困惑,例如当您尝试验证基因的起始蛋氨酸但 blastx 结果在更远的氨基酸处开始比对时。我们始终在未选中“低复杂性区域”过滤器的情况下运行我们的blastx 搜索,以便将这些区域包含在搜索中以最大化对齐长度。
省时提示#2:blastx 搜索本质上比blastn 或blastp 慢,因为在将核苷酸序列翻译成所有六个可能的阅读框时需要进行额外的搜索。如果您知道预期的蛋白质序列,请使用“对齐两个或更多序列”选项来大幅减少搜索结果的等待时间。
优化blastp搜索
根据测序结果,我们经常在标准蛋白质 BLAST (blastp)和blastx 搜索之间进行选择,以验证质粒中的预期蛋白质序列。如果您知道为测序结果选择哪个阅读框并且可以轻松翻译它,我们建议您使用blastp 而不是blastx。主要优点是节省时间,但另一个好处是,默认情况下,blastp 搜索不会过滤低复杂度区域,这意味着您不必记住调整任何blastp 算法参数。我们使用默认评分矩阵 BLOSUM62,但您可能需要检查其他矩阵的描述,看看其他矩阵是否对您的搜索更有利。
省时提示#3:请注意,可用的蛋白质数据库不太可能为您最喜欢的基因与表位标签或融合蛋白融合提供准确的条目。如果选择您的测序引物来确认标签或融合蛋白符合读框,我们建议使用带有“对齐两个或更多序列”选项的blastx,并将您预期的蛋白质序列粘贴到“主题序列”框中。
BLAST 替代品
根据您的测序结果和所需的分析,BLAST 可能并不总是您的最佳选择。对于 BLAST 无法处理的困难序列比对,Clustal是我们对核苷酸或蛋白质序列进行成对或多序列比对的常用选择。我们还使用COBALT来比对多个蛋白质序列,特别是用于比较不同的同种型。除了我们最喜欢的之外,还有许多可用的序列比对工具。
尝试这些资源以获取 BLAST 的替代品列表:
ExPASy - http://www.expasy.org/genomics/sequence_alignment
EMBL-EBI - http://www.ebi.ac.uk/services
http://www.ebi.ac.uk/Tools/webservices/#multiple_sequence_alignment_msa
参考资料
