【4.2】机器学习在药物发现和开发中的应用
今早,葛博在群里推荐了这篇综述。光是这个期刊,这个文章名,就值得很认真的读一下这个文献。
药物发现和开发管道(pipelines)漫长,复杂并且取决于许多因素。机器学习(ML)方法提供了一套工具,可以通过丰富、高质量的数据改善指定问题的发现和决策。应用ML的机会发生在药物发现的所有阶段。实例包括靶标验证,预后生物标记物的鉴定和临床试验中数字病理学数据的分析。应用程序的范围和方法不同,有些方法可以产生准确的预测和见解。应用ML的挑战主要在于ML产生的结果缺乏可解释性和可重复性,这可能限制其应用。在所有领域,仍然需要生成系统和全面的高维数据。通过不断努力解决这些问题,以及提高对验证ML方法所需因素的认识,ML的应用可以促进数据驱动的决策,并有可能加快流程并降低药物发现和发展。
生物系统是发育和疾病期间复杂的信息根源。 现在,使用大量的“组学”和智能技术,系统地测量和挖掘这些信息,达到前所未有的水平。 这些高通量生物学和疾病方法的出现为制药行业带来了挑战和机遇,其目的是确定开发药物的合理治疗假设。 然而,许多因素的最新进展已导致对制药工业中机器学习(ML)方法的使用的兴趣增加。 再加上无限可扩展的存储,可以为ML提供基础的数据集类型和数据大小的大量增加,使制药公司能够访问和组织更多数据。 数据类型可包括图像,文本信息,生物识别和可穿戴设备的其他信息,化验信息和高维组学数据。
在过去几年中,人工智能(AI)领域已从大量的理论研究转向实际应用。 这种爆炸性增长大部分与图形处理单元(GPU)等新计算机硬件的广泛可用性有关,这使得并行处理更快,尤其是在数字密集型计算中。 最近,新的ML算法的进步,例如深度学习(DL)2,从数据构建强大的模型,以及这些技术在众多公共竞赛中的可证明的成功,在过去2年有助于极大地增加ML在制药公司中的应用 。
尽管许多消费者服务行业早已采用ML领域的新方法,但制药行业的采用率一直滞后于最新。 众所周知,药物开发的成功率(从第一阶段临床试验到药物批准的定义)在所有治疗领域和整个制药行业都非常低。 最近对21,143种化合物的研究发现,总体成功率低至6.2%。 因此,制药行业中使用ML技术的许多理由是由业务需求驱动的,以降低总体损耗和成本。
药物发现和开发的所有阶段,包括临床试验,已着手开发和利用ML算法和软件(图1)来识别新靶点,为目标疾病关联提供更有力的证据,改进小分子化合物的设计和优化,增加了解疾病机制,增加对疾病和非疾病表型的理解,为预后、进展和药物疗效开发新的生物标志物,改进患者监测和可穿戴设备的生物识别和其他数据分析,增强数字病理成像并从各级分辨率的图像中提取高内容信息。
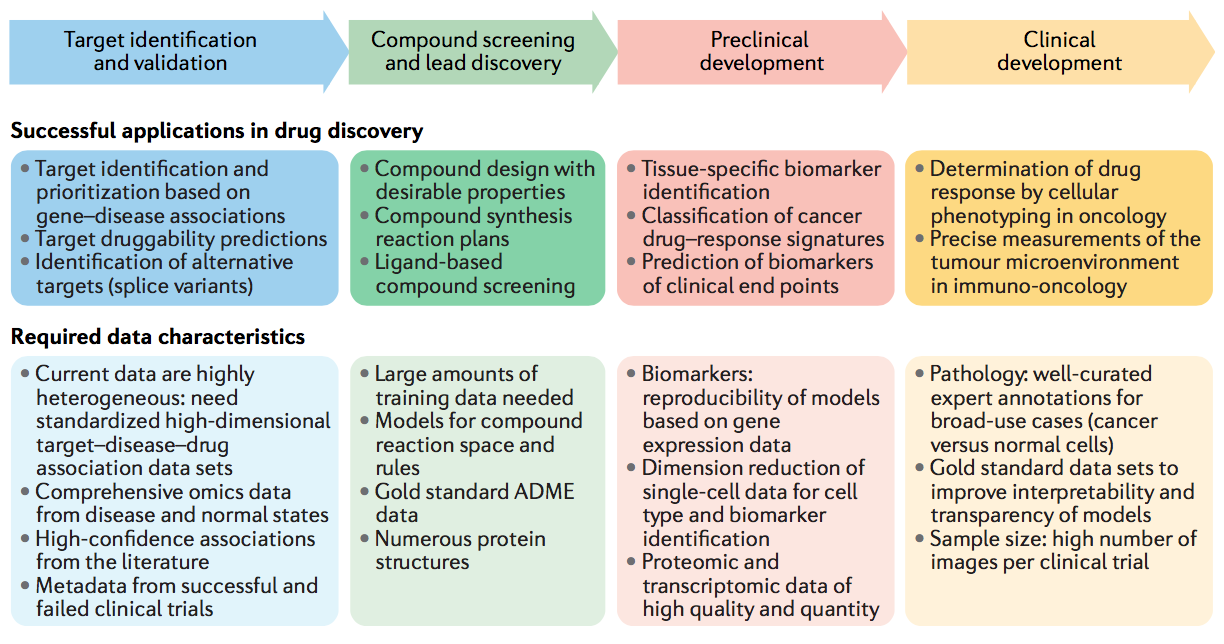
图1 | 药物发现管道中的机器学习应用程序及其所需的数据特征。 已经出版了几种在制药公司药物开发管道的各个阶段中机器学习的成功应用。 但是,在每个数据域内,仍然存在与数据质量标准和数据量相关的挑战,以充分利用这些发现方法的潜力。 ADME,吸收,分布,代谢和排泄(absorption, distribution, metabolism and excretion)。
因此,许多制药公司已开始投资于资源,技术和服务,生成和策划数据集,以支持该领域的研究。 此外,IBM和谷歌等技术巨头,生物技术初创企业和学术中心不仅提供基于云的计算服务,还与行业合作伙伴一起在制药和医疗保健领域工作。 本综述概述了ML中使用的当前工具和技术(工具箱),包括深度神经网络,以及迄今为止在关键药物应用领域的进展概述。
一、机器学习工具箱
从根本上说,ML是使用算法解析数据,从中学习数据然后对任何新数据集的未来状态做出决定或预测的实践。 因此,不是手工编写具有特定指令集(由程序员预先确定)来完成特定任务的软件例程,而是使用大量数据和算法对机器进行训练,使其能够学习如何执行 任务。 程序员编码用于训练网络的算法而不是编码专家规则。
随着可用于学习的数据的数量和质量的增加,算法自适应地改善其性能。 因此,ML最适用于解决大量数据和若干变量即将出现的问题,但与这些问题相关的模型或公式尚不清楚。
有两种主要类型的技术用于应用ML:监督和无监督学习。 监督学习方法用于开发训练模型以预测数据类别或连续变量的未来值,而无监督方法用于探索目的以开发能够以用户未指定的方式聚类数据的模型。 监督学习训练已知输入和输出数据关系的模型,以便它可以预测新输入的未来输出。 未来的输出通常是数据分类的模型或结果,或者是对最有影响的变量(回归)的理解。 无监督学习技术识别输入数据中的隐藏模式或内在结构,并使用它们以有意义的方式聚类数据。
1.1 模型选择概念
良好的ML模型的目的是从训练数据到手头的测试数据很好地概括。 泛化(Generalization)指的是模型学习的概念在训练期间应用于模型未见的数据的程度。 在每种技术中,存在几种方法(图2),它们的预测准确度,训练速度和它们可以处理的变量数量各不相同。 必须仔细选择算法,以确保它们适合于手头的问题以及可用数据的数量和类型。 所需的参数调整量以及该方法将信号与噪声分离的程度也是重要的考虑因素。
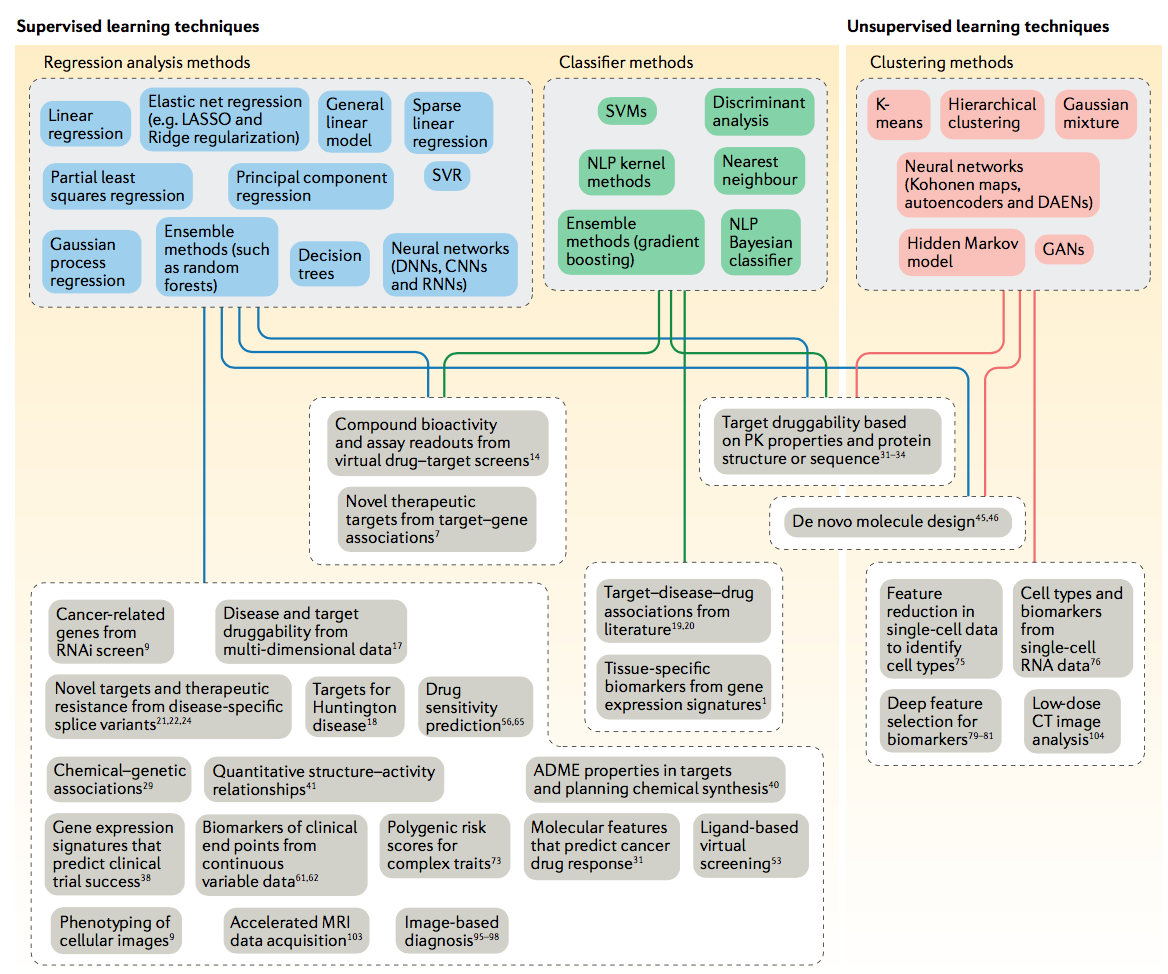
图2 | 机器学习工具及其药物发现应用。 该图概述了用于回答本评价中涉及的药物发现问题的机器学习技术。 一系列监督学习技术(回归和分类器方法)用于回答需要预测数据类别或连续变量的问题,而无监督技术用于开发能够实现数据聚类的模型。 ADME,吸收,分布,代谢和排泄; CNN,卷积神经网络; CT,计算机断层扫描; DAEN,深度自动编码器神经网络; DNN,深度神经网络; GAN,生成对抗网络; MRI,磁共振成像; NLP,自然语言处理; PK,药代动力学; RNAi,RNA干扰; RNN,递归神经网络; SVM,支持向量机; SVR,支持向量回归。
当模型不仅学习信号而且学习训练数据的一些不寻常特征并将这些特征结合到模型中时,模型过度拟合(overfitting)发生,从而对模型在新数据上的性能产生负面影响。欠拟合(Underfitting)指的是既不能对训练数据建模也不能推广到新数据的模型。限制过度拟合的典型方法是应用重采样方法或阻止部分训练数据用作验证数据集。随着模型复杂性的增加,正则化回归方法(如Ridge,LASSO或elastic nets)会增加参数的惩罚,从而迫使模型generalize数据而不是过度拟合。避免过度拟合的最有效方法之一是dropout方法,它随机删除隐藏层中的单位。不同的ML技术具有不同的性能指标。基本评估指标,例如分类准确度,kappa,曲线下面积(AUC),对数损失,F1分数和混淆矩阵可用于比较各种方法的性能。黄金标准数据集的可用性以及独立生成的数据集在生成性能良好的模型中非常有用。
现在有几个软件库可用于跨各种硬件平台(中央处理单元(CPU),GPU和张量处理单元(TPU))以及从台式机到服务器集群的高性能数学计算。 常用的ML程序框架是开源框架TensorFlow,最初由Google AI组织中的Google Brain团队的研究人员和工程师开发,以及PyTorch,Keras和Scikit-learn。
1.2 深度神经网络架构 (Deep neural network architectures )
DL是20世纪80年代和90年代的人工神经网络的现代转世,它使用复杂的多级深度神经网络(DNN)来创建可以从大量未标记或标记的训练数据中执行特征检测的系统。 DL与传统人工神经网络的主要区别在于所用网络的规模和复杂性。在神经网络中,输入特征被馈送到输入层,并且在使用隐藏层的多个非线性变换之后,预测由输出层生成。这通常通过使用误差的反向传播来逐步减小所获得的和输出的预期值之间的差异来完成。每个输出节点对应于要预测的任务(或类)。如果输出层中只有一个节点,则相应的网络称为单任务神经网络。 DL可以拥有大量隐藏层,因为它使用更强大的CPU和GPU硬件,而传统神经网络由于硬件限制通常使用一个或两个隐藏层。 DL中还有许多算法改进。
DNN在药物发现中的应用很多,包括生物活性预测,从头分子设计,合成预测和生物图像分析。 DNN的一个优点是它们具有下面描述的几种不同的灵活架构,因此用于回答各种问题:
- 在第一种体系结构中,深度卷积神经网络(CNN),一些隐藏层仅在本地(而不是全局)连接到下一个隐藏层。 CNN通过将简单的局部特征分层组合成复杂模型,在语音和图像识别等领域实现最佳预测性能。图形卷积网络是一种特殊类型的CNN,可以以图形或网络的形式应用于结构化数据。
- 第二种体系结构是递归神经网络(RNN),其采用神经网络的重复模块链的形式,其中节点之间的连接形成沿序列的有向图。这允许分析需要持久信息的随时间的动态变化。长期短期记忆神经网络是一种特殊的RNN,能够学习长期依赖性。
- 第三个例子 - 完全连接的前馈网络 - 是每个输入神经元连接到下一层中每个神经元的网络。这与RNN相反,在完全连接的前馈网络中,梯度是通过反向传播明确定义和计算的。这些模型已用于具有挑战性的预测模型构建案例,例如基因表达数据,其中样本数量相对于特征数量较小。
- 第四种网络架构是深度自动编码器神经网络(DAEN)。这种类型的神经网络是一种无监督学习算法,它应用反向传播将其输入投影到其输出,以降低尺寸,从而试图保留数据的重要随机变量,同时去除非必要部分。
- 第五个也是最后一个网络架构 - 生成对抗网络(generative adversarial net- works,GAN) - 由任意两个网络组成(尽管通常是前馈神经网络和CNN的组合),其中一个负责生成内容,另一个负责对内容进行分类。
1.3 数据特征
据说ML的实践包括至少80%的数据处理和清理以及20%的算法应用。因此,任何ML方法的预测能力取决于高质量的大量数据的可用性。用于培训的数据需要准确,精确并尽可能完整,以便最大限度地提高可预测性。实验设计通常涉及关于理想样本大小的讨论以及用于正确估计该参数的适当功率计算。是否可以获得正确类型的数据以及应该通过实验生成哪些数据也是某些问题的关键考虑因素。当用于以系统方式生成的数据时,ML应用程序更加强大,具有最小的噪声和良好的注释。如下所述,许多应用程序并不是特别有效,因为数据是从具有可变数据质量的多个来源组合而来的。目前正在努力开发药物发现的特定领域中的开放注释数据,例如目标验证。这些目标旨在在对药物发现和开发具有重要意义的领域产生高质量的正面和负面注释,以促进ML的应用。
二、在药物发现中的应用
2.1 目标识别和验证。
药物发现中的卓越方法是开发药物(小分子,肽,抗体或更新的方式,包括短RNA或细胞疗法),通过调节分子靶标的活性来改变疾病状态。尽管最近表型筛选再次出现,但是启动药物开发计划需要使用合理的治疗假设来鉴定靶标:靶标的调节将导致疾病状态的调节。根据现有证据选择该目标称为目标识别和优先级。做出这个初步选择后,下一步是使用生理学相关的离体和体内模型(目标验证)验证所选靶标在疾病中的作用。尽管目标的最终验证只会在稍后进行,但通过临床试验,早期目标验证对于将重点放在可能成功的项目上至关重要。
现代生物学的数据越来越丰富。 这包括大群体中的人类遗传信息,健康个体的转录组学,蛋白质组学和代谢组学谱分析以及具有特定疾病和临床材料的高含量成像的。 捕获这些大型数据集,并通过公共数据库重用它们的能力为早期目标识别和验证提供了新的机会。 然而,这些多维数据集需要适当的分析方法来产生统计上有效的模型,这些模型可以对目标识别进行预测,这就是可以利用ML的地方。 可以有助于目标识别和验证的实验范围很广,但如果这些实验是数据驱动的,则ML正在被越来越多地应用。
目标识别的第一步是建立目标与疾病之间的因果关联。建立因果关系需要证明目标的调节会影响自然发生(遗传)变异或精心设计的实验干预的疾病。然而,ML可以用于分析具有关于假定目标的功能的信息的大数据集,以预测潜在的因果关系,例如,由已知真实目标的属性驱动。已经以这种方式在目标识别领域的若干方面应用了ML方法。 Costa等人建立了一个基于决策树的元分类器,其培训蛋白质蛋白质的网络拓扑,代谢和转录相互作用,以及组织表达和亚细胞定位,以预测与发病率相关的基因,这些基因也是可药物的。通过检查决策树,他们确定了多种转录因子(TF)的调节,代谢途径的中心性和细胞外位置作为关键参数。在其他研究中,ML模型专注于特定疾病或治疗领域。 Jeon等人使用各种基因组数据集构建了支持向量机(SVM)分类器,将蛋白质分类为药物靶标和乳腺癌,胰腺癌和卵巢癌的非药物靶标。关键分类特征是基因必需性,mRNA表达,DNA拷贝数,突变发生和蛋白质 - 蛋白质相互作用网络拓扑。总共确定了122个全球癌症靶点,其中69个与116个已知癌症靶点重叠。此外,266,462和355个目标被确定为分别对乳腺癌,胰腺癌和卵巢癌具有特异性。使用在细胞培养模型中具有强抗增殖作用的肽抑制剂验证了两个预测的靶标。此外,137种预测的胰腺癌靶标的抑制剂几乎是其他化合物显示出对细胞活力的强烈抑制的两倍。 Ament等人建立了一个基于小鼠TF结合位点和转录组分析数据的模型,以表征亨廷顿病的转录变化。他们使用回归模型和LASSO正规化重建了小鼠纹状体中718个TF的靶基因的基因组规模模型。总体而言,48个已鉴定的TF模块中的13个在人类疾病的纹状体组织中差异表达,并为亨廷顿病治疗提供了潜在的起点。 Mamoshina等人已经鉴定了组织特异性抗衰老疗法的分子靶标。他们比较了年轻和年老肌肉的基因表达特征。几种有监督的ML方法的比较揭示了具有线性核和深度特征选择的SVM最适合于老化生物标志物的鉴定。在这些例子的每一个中,ML产生了一组目标预测,这些目标具有表明它们可能与药物结合或参与疾病的特性,但进一步验证对于产生治疗假设至关重要。
文献是目标与疾病相关的知识的主要来源。 文献的自动处理解锁了非结构化文本中的信息,否则这些信息将无法访问。 自然语言处理(NLP)(一种应用于文本挖掘的ML方法)的最新进展使得更有效的数据挖掘能够识别相关论文。 BeFree应用NLP核心方法来识别Medline摘要中的药物 - 疾病,基因 - 疾病和靶 - 药物关联。 这种监督学习方法依赖于手动注释的欧盟不良药物反应(EU-ADR)数据库关系语料库和基于遗传关联数据库的半自动注释语料库。 DigSee在Medline摘要中识别基因和疾病,使用NLP提取这些实体之间的生物事件,并使用贝叶斯分类器对证据句子进行排序。
ML的一个广阔领域是了解生物学的基本方面,通过替代方式或新目标确定治疗机会。 了解剪接信号中的遗传变异就是一个例子。 DL拼接模型现在能够准确地预测交替拼接信号。 最新的整合剪接模型将体内剪接因子结合的CLiP-seq测定数据与RNA测序实验相结合,其中这些剪接因子已经被敲低或过表达。 将剪接代码模型与从头和复杂剪接变异的预测相结合,可以鉴定阿尔茨海默病特异性的剪接变体。 最近类似方法的应用确定了CART-19免疫疗法的逃避机制,导致耳聋的罕见遗传变异和与孤独症相关的剪接变异。
ML还可以预测癌症特异性药物效应。 Iorio等人针对265种抗癌药物筛选了990种癌细胞系,并研究了全基因组基因表达,DNA甲基化,基因拷贝数和体细胞突变数据如何影响药物反应。他们使用ANOVA,逻辑模型和ML算法(弹性网络回归和随机森林)来识别预测药物反应的分子特征。癌症类型中最具预测性的数据类型是基因表达,而最具预测性的癌症特异性模型包括基因组特征(驱动突变或拷贝数改变),如果它们包括DNA甲基化数据则甚至更好。 Tsherniak等人使用来自501个癌细胞系的RNA干扰(RNAi)筛选的数据来找到预测769个基因的癌症依赖性的分子标记。他们开发了一种基于条件推理树的非线性回归模型,以生成基于基因表达,基因拷贝数和体细胞基因突变的预测模型。 McMillan等人对超过100个重复注释的多种和特征性躯体肺癌病变的细胞模型筛选了222种化学物质。他们应用正则化的ML( elastic net )和基于概率的度量(扫描Kolmogorov-Smirnov)来识别171种化学 - 遗传关联,这些关联揭示了在没有有效治疗的一系列癌症类型中可靶向的机制脆弱性。这些方法表明存在肿瘤内在精准医学的机会。
对于药物开发者来说,另一个重要问题是药物可以用于任何给定目标的可能性有多大。对于小分子药物,这需要鉴定具有暗示这些蛋白质可以结合小分子的特征的靶标。可以使用不同的目标属性来生成这些药物可行性模型。 Nayal和Honig根据一组99种蛋白质对99种药物结合和1,187种非药物结合腔的物理化学,结构和几何属性进行了随机森林分类。表面腔的尺寸和形状是最重要的特征。一些研究从已知药物和非药物靶标的蛋白质序列衍生出各种物理化学特性,并应用具有堆叠自动编码器(DL模型)的SVM或偏向SVM来预测可药物靶标。还发现可药用蛋白质占据蛋白质蛋白质相互作用网络的特定区域并且倾向于高度连接。同样,这些ML方法的例子产生了预测可能与药物结合的目标组,因此减少了潜在的搜索空间,但这些目标需要进一步验证。
用于目标识别或验证的圣杯是,对基于目标的药物发现计划的未来临床试验成功的早期预测。各种非ML分析指出了成功的可能预测因素。使用ML,Rouillard等人通过多变量特征选择评估了一组332个目标的组学数据,这些目标通过III期临床试验成功或失败。他们发现基因表达数据特别能预测成功的靶点,其特征在于低平均RNA表达和跨组织的高变异。该研究证实了先前的发现,即理想靶标在受影响的组织中表现出疾病特异性表。Ferrero等人使用来自开放目标平台的目标疾病关联训练了一系列ML分类器,以预测从头潜在的治疗目标。对特征重要性的评估确定了动物模型,基因表达和遗传数据的存在作为治疗目标预测的关键数据类型,而与指示无关。但是,这种方法受到数据稀疏性和缺乏启动程序失败原因信息的限制。更根本的是,由于启动成功的药物发现计划和将药物推向市场之间的时间长,成功的计划反映了早期药物开发的范例。成功的小分子计划的驱动力今天不太可能是相同的,因为新的形式可用,如生物制剂(包括抗体)。对精准医学的日益关注引入了额外的限制。对于未来的预测方法而言,关于成功和失败的药物发现计划的广泛数据,可用于公共领域的元数据是至关重要的。
2.2 小分子设计和优化
可以阻断或激活目标靶蛋白的候选药物的发现,涉及对大化合物文库的广泛的虚拟和实验高通量筛选。 然后进一步精制和修饰候选结构以改善靶特异性和选择性,以及优化药效学、药代动力学和毒理学性质。 但重要的是,缺乏足够的高质量数据用于新化学,如蛋白水解 - 靶向嵌合体(PROTAC)和大环化合物,可能会限制ML对这种化学反应的影响。
已经做了很多工作来将DL方法(例如多任务神经网络)应用于基于配体的虚拟筛选。给定lead化合物,可以通过计算方法鉴定具有相似化学结构的化合物。这通常使用经典的统计方法来执行,但多任务DNN被证明更有效。当推断小分子的性质和活性时,DNN可以显着提高预测能力。一次性学习技术可用于显着减少在新实验装置中对分子读数进行有意义预测所需的数据量。将ML与马尔可夫状态模型相结合,该技术用于鉴定opiate与 μ-opioid receptor结合的先前未知机制,揭示了与其活化有关的变构位点。然而,多任务模型相对于单任务模型的好处是高度依赖数据集的。为了帮助对ML算法进行基准测试,Pande等人编制了一个大型基准数据集MoleculeNet,该数据集已用于比较不同的ML算法。 MoleculeNet包含超过700,000种化合物的性质数据。所有数据集都已经过策划并集成到开源DeepChem软件包中(参见相关链接),其中还包括其他工具。
DNN和现代树搜索算法( modern tree search algorithms )也可用于规划化学合成的有效路线。为了计划靶分子的合成,使用逆反应(反向合成)将分子正式分解。该过程产生一系列反应,然后可以在实验室中在向前方向上执行以合成目标。一个主要挑战是系统地将合成化学知识应用于该过程。由于化学知识呈指数增长,并且许多反应的范围和局限性尚未完全理解,因此手动纳入转换规则是令人望而却步的。为了自动提取规则,Segler等人使用Reaxys数据库(大约1100万个反应和~300,000个规则)并执行蒙特卡罗树搜索(MCTS)以与DNN一起对树节点进行评分,从而引导最有希望的搜索方向。在定量分析中,该方法优于金标准,最佳第一次搜索,具有两种不同的实现(启发式方法和神经)。此外,对于近三分之二的检测分子,MCTS比传统的计算机辅助搜索方法快30倍。在双盲研究中也进行了定性测试。有机化学家被要求在不知道如何获得路线的情况下在基于文献和预测的合成路线之间进行选择。在这里,化学家们第一次认为预测路线的质量平均与从文献中获得的路线一样好。
DL的另一个有价值的应用是通过强化学习进行分子从头设计。 阿斯利康的研究人员利用RNN扩展化学空间,通过调整基于序列的生成模型来设计具有几乎最佳溶解度,药代动力学特性,生物活性和其他参数值的化合物。 Kadurin等人还开发了类似的模型,使用深GAN在非常大的数据集上进行分子特征提取。但是,必须指出的是,强化学习可能无助于确定新的和前所未有的合成路线。
社区问题解决竞赛可以用于推进特定领域的方法开发。 Merck Sharp&Dohme的研究人员赞助了一项Kaggle比赛,用于预测其他相关的吸收,分布,代谢和排泄(ADME)参数以及一些生化目标。获奖团队使用DNNs,在15个测定系统中的13个中,其表现略好于标准随机森林。他们的一些关键学习是超参数的优化可以改善DNN,不需要特征选择,多任务模型比单任务模型表现更好,并且可以通过使用dropout来防止过度拟合。 Ramsundar等人还观察到多任务DNN的性能优于单任务DNN。 Lenselink等人使用从ChEMBL获得的一个标准化数据集来追踪单任务和多任务DNN之间的比较以及不同ML方法(随机森林,SVM,朴素贝叶斯和逻辑回归)之间的比较。在这里,DNN模型表现最佳,并且还发现多任务DNN优于单任务DNN。多任务DNN也被证明对于lead优化和铅识别的预测更好,因为它们可以由于输出层中存在多个节点而从许多不同的生物源合成信息。
如Kramer和Gütlein的研究所示,模型构建前的特征选择可以改善ML模型。 他们还能够检测随机森林模型与其他ML方法(如SVM和朴素贝叶斯)的改进,在训练模型时性能更快,使用的功能更少。 在他们看来,过滤化学指纹位的一个主要好处是模型可解释性的提高。 如果未过滤指纹,则由于称为“比特碰撞(bit collisions)”的影响,可解释性受到阻碍。 Landrum等人也独立地显示了过滤指纹的关键影响
Hochreiter等人还发现,基于DNN的模型明显优于所有竞争方法,并且使用所有ChEMBL分析的数据集和基于简化分子输入线进入系统(SMiLES)输入的目标预测,DL的预测性能在 许多案例与在湿实验室进行的测试相当。 Hochreiter小组还表明,DNNs优于所有其他ML方法(k-最近邻,朴素贝叶斯,随机森林和SVM)和基于统计的方法(相似性方法53)用于目标预测。 同一组赢得了2014年Tox21数据挑战赛的大部分挑战。
小分子设计领域尚未解决的挑战是如何最好地代表化学结构。 存在大量的表示,从简单的圆形指纹,例如扩展连通性指纹(ECFP)到复杂的对称功能(图3)。 目前尚不清楚哪种结构表示最适合哪种小分子设计问题。 因此,有趣的是,在化学信息学领域ML研究的增加是否会为结构表征的最佳选择提供更多指导。
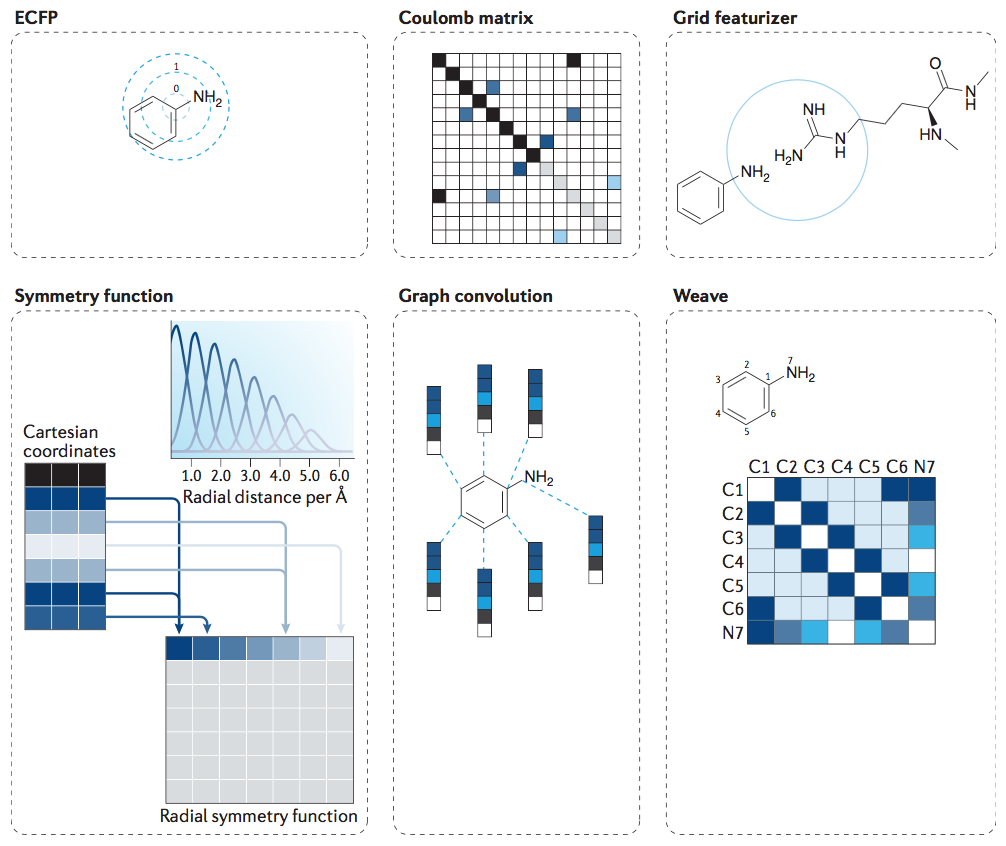
图3 复合结构表示在机器学习模型中的挑战。化学结构及其特征的适当表示可以根据所需的应用进行多种表示。扩展连接指纹( Extended-connectivity fingerprints ,ECFP )包含有关分子拓扑特征的信息,可以将此信息应用于相似性搜索和活动预测等任务。 库仑矩阵编码有关分子核电荷及其坐标的信息。网格特征化方法结合了配体和靶蛋白的结构特征以及有助于结合亲和力的分子间力。对称函数是原子坐标信息的另一种常见编码,它关注原子对之间的距离和原子三重态内形成的角度。图卷积方法计算每个原子的初始特征向量和邻居列表,总结原子的局部化学环境,包括原子类型,杂化类型和价结构。编织特征计算分子中每对原子的特征向量,包括键属性(如果直接连接),图形距离和环信息,形成特征矩阵。(Wu, Z. et al. MoleculeNet:abenchmarkformolecularmachinelearning.Chem.Sci.9,513–530(2018))
2.3 预测性生物标志物
基于ML的生物标志物发现和药物敏感性预测模型,被证明是有助于提高临床成功率,更好地了解药物作用机制以及为正确患者确定正确药物的方法。 后期临床试验需要花费许多年和数百万美元才能进行,因此使用临床前和/或早期临床试验数据建立,验证和应用预测模型将是最有益的。 可以使用ML方法在临床前数据集上预测翻译生物标志物。 在使用独立数据集(临床前或临床)验证后,该模型及其相应的生物标志物可用于对患者进行分层,确定潜在适应症并建议药物作用机制(图4)
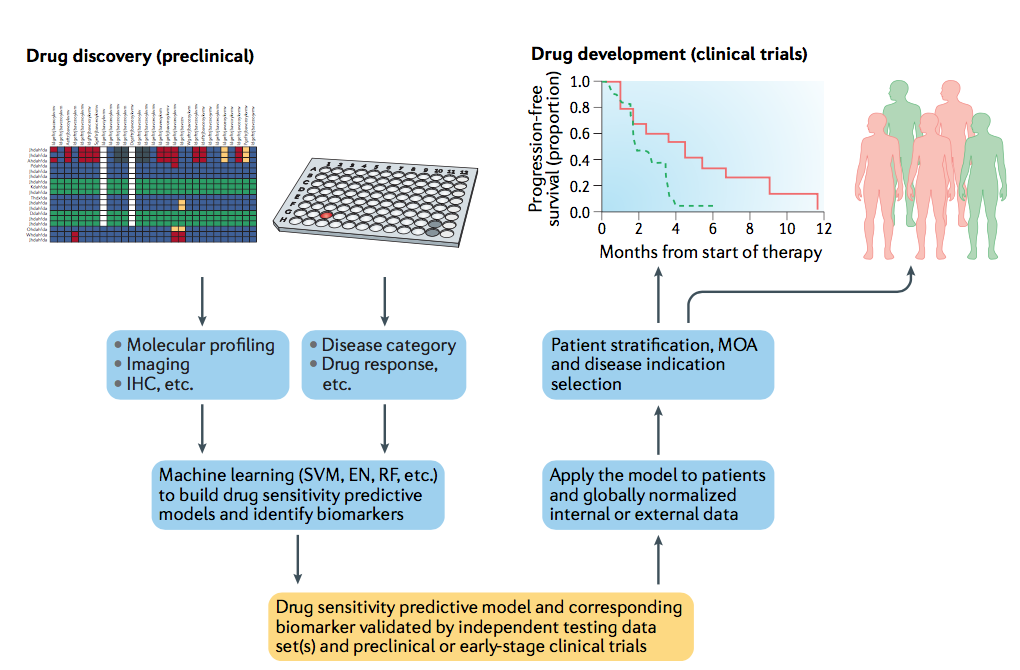
图4 | 利用预测生物标志物来支持药物的发现和开发。 可以使用关于临床前数据的机器学习方法生成药物敏感性预测模型(黄色框)。 然后可以使用来自早期临床患者样品的数据测试该模型。 一旦经过验证,该模型可用于患者分层和/或疾病指征选择,以支持药物的临床开发,以及推断其作用机制。 EN,弹性网; IHC;免疫组化; MOA,行动机制; RF,,随机森林; SVM,支持向量机。
尽管文献中有数千篇关于生物标志物和预测模型的论文,但其中很少有用于临床试验。各种因素导致了这一差距,包括数据质量,模型选择,数据和软件访问,模型再现性以及适合临床环境的检测设计。为了解决一些与模型相关的问题,一些社区工作已经评估了ML方法来开发分类和回归模型。几年前,美国食品和药物管理局(FDA)组织了MicroArray质量控制II(MAQC II)计划,以评估各种ML方法,用于预测基线基因表达数据的临床终点。在该项目中,36个独立团队分析了6个微阵列数据集,以生成预测模型,用13个临床终点中的1个对样本进行分类。一般观察包括数据质量控制过程的重要性,熟练科学家的需求(一些团队的表现始终优于使用相同ML方法的其他团队)以及为临床终点选择合适的建模方法的重要性。例如,对多发性骨髓瘤患者的总体生存率预测不佳可能部分是由于应用了24个月的任意生存期限。多发性骨髓瘤中的基因表达和总体存活都是连续变量,因此,基于回归的预测模型是合适的。实际上,使用单变量Cox回归方法,确定了显着预测高风险亚组患者的基因表达特征。这个标签在几个独立研究和不同的基于回归的方法中得到了证实,突出了没有预定义类成员资格的回归方法的优势
国家癌症研究所(NCI)-DREAM挑战是另一项社区努力,旨在评估建立药物敏感性预测模型的回归方法(定义为回归问题)。每个参与团队使用他们最好的建模方法并在相同的训练数据集(用31种药物治疗的35个乳腺癌细胞系)上优化他们的参数集,然后在相同的盲法测试数据集上测试他们的模型的性能(18乳腺癌)细胞系用相同的31种药物治疗)。六种类型的基线分析数据可用于生成预测模型 - RNA微阵列,单核苷酸多态性(SNP)阵列,RNA测序,反相蛋白质阵列,外显子组测序和DNA甲基化状态 - 44个参与团队应用各种回归方法如核方法,非线性回归(回归树),稀疏线性回归,偏最小二乘回归,主成分回归或集合方法。与MAQC II结果一致,一些团队使用相同的方法始终优于其他团队。差异性能可能反映了用于质量控制,数据简化,特征选择,分裂策略和微调ML参数的技术细节,以及潜在的生物学知识结合,如基因功能信息或临床将数据纳入预测模型的构建中。此外,对于所有团队和方法,某些药物比其他药物更容易构建预测模型。 NCI-DREAM挑战数据集和结果继续用作方法开发和评估的验证数据集,例如,新的随机整体集合框架,群体因子分析和其他方法
现已发表了几个成功的案例研究,其中ML生成的预测模型及其相应的生物标记物在药物发现和开发中发挥了关键作用。 Li等人使用标准护理药物进行了一项案例研究,其中他们首先使用癌细胞系筛选数据建立了对厄洛替尼和索拉非尼(每种药物的一种模型)的药物敏感性模型。然后,他们应用这些模型对来自BATTLE临床试验的患者进行分层,这些患者接受了两种药物中的一种治疗,并证明这些模型具有预测性和药物特异性。模型衍生的生物标记基因显示反映了每种药物的作用机制,当与来自各种癌症类型的全球标准化公共领域数据相结合时,该模型预测了癌症类型对每种药物的敏感性,这与其FDA一致批准的适应症。该研究表明,使用ML方法以组织无关的方式识别导致各种癌症类型的药物敏感性的关键特征可能对药物开发有用(与基于癌症类型的临床试验相比,随后是标签扩展)。 2017年,FDA批准了程序性细胞死亡1(PD1)抑制剂pembrolizumab用于具有特定遗传生物标志物的癌症。这是基于交叉适应症遗传生物标志物而非癌症类型的第一个FDA批准,突出了对更多基于机制的生物标志物发现的需求。
最近,在使用各种类型的输入数据的肿瘤学以外的适应症中,基于ML的预测生物标志物已经取得了很大进展。 Tasaki等人将ML方法应用于多组学数据,以更好地了解类风湿性关节炎患者的药物反应。 Pare等人开发了一种基于梯度增强回归树的新型ML框架,用于构建预测复杂性状的多基因风险评分。 在英国生物银行数据集上进行测试,他们的基于SNP的模型能够分别解释高度和BMI的总体多基因方差的46.9%和32.7%。 此外,Khera等人开发了全基因组多基因评分,以鉴定具有冠状动脉疾病,心房颤动,2型糖尿病,炎性肠病和乳腺癌高风险的个体。
单细胞RNA测序技术的快速发展已被用于基因聚类和细胞特异性生物标志物发现。已经使用单细胞RNA测序技术来鉴定新细胞类型,区分细胞状态,追踪发育谱系并将表达谱与细胞的空间分辨率整合。然而,未解决的挑战是基因表达测量从数万个细胞减少到低维空间,通常是两个或三个变量。 Ding等人开发了一种概率生成模型scvis,用于在具有不确定性估计的单细胞基因表达数据中将高维空间减少到低维结构。然后该工具用于分析四个单细胞RNA测序数据集并产生多维单细胞RNA测序数据的2D表示,其可被解释为稳健地鉴定细胞类型。此外,Rashid等人使用变异自动编码器(VAE)将单细胞RNA测序数据转换为潜在编码特征空间,以更有效地区分隐藏的肿瘤亚群。对编码特征空间的分析揭示了细胞亚群及其之间的进化关系。该方法完全没有监督,并且需要对数据进行最少的预处理。另外,该方法耐受单细胞RNA测序数据集中的基因表达缺失。 Wang和Gu提出了用于单细胞RNA测序数据(VASC)的深变分自动编码器,这是一种深层多层生成模型,用于无监督的降维和可视化数据。经过对20个数据集的测试,VASC具有优越性,并且具有比ZIFA和SIMLR等几种最先进的降维方法更广泛的数据集兼容性
ML最近的一个令人兴奋的发展是生物标志物发现的特征选择的迅速增加。 例如,研究人员应用无监督的DL模型来提取基因模块或样本群的有意义的representations。 Way和Greene介绍了在癌症基因组图谱(TCGA)泛癌RNA测序数据上训练的VAE模型,并鉴定了VAE编码特征中的特定模式。 Beck等人进行了基因表达和蛋白质组学数据的图像分析和数据整合,以改善肺鳞状细胞癌的鉴定。 Nirschl等人表明,CNN模型可以比两位训练有素的心脏病学家(AUC = 0.73和0.75)更好地预测心内膜心肌活检样本发生心力衰竭的可能性(AUC = 0.97)。
在所有这些例子中,对于ML生成的预测生物标记物更成功,仍有几个关键问题需要解决。 这些问题中的至少一些涉及分类器的可解释性,至少一些最终用户认为这对于临床采用是至关重要的。 其他一个关键问题是需要在多站点,多机构数据集的背景下验证这些方法,以证明该方法的普遍性。 研究界正在积极解决这些问题并取得快速进展,包括应用模型训练和参数优化的客观方法和措施,模型解释和生物学见解的提取,以及模型再现性
2.4 计算病理学
病理学(pathology)是一个描述性领域,因为病理学家通过视觉检查来解释在载玻片上看到的内容。 对这些载玻片的分析提供了大量信息,例如组织中存在的细胞类型及其空间背景。 肿瘤微环境中肿瘤和免疫细胞之间的相互作用在免疫肿瘤学研究中越来越重要,并未被其他技术所捕获。
制药公司需要了解药物治疗如何影响特定组织和细胞,并且需要在选择临床试验候选者之前测试数千种化合物。 此外,随着临床试验数量的增加,发现新的生物标志物对于识别对特定疗法有反应的患者将变得越来越重要。 增加使用计算病理学可以允许发现新的生物标记物并以更精确,可重复和高通量的方式产生它们将最终减少药物开发时间并允许患者更快地获得有益疗法。
在DL之前,用于组织图像分析的算法通常与病理学家合作得到生物学启发,并且需要计算机科学家为计算机手工描述特征以对特定类型的组织或细胞进行分类。这些研究旨在识别广泛使用的血红素和曙红(haemotoxylin and eosin,H&E)染色图像中的形态描述符。核形态测定法是最早的计算病理学实施之一,证明了确定计算机生成的特征与预后之间关联的能力。 Beck等人研究了周围肿瘤基质内空间位置的细胞,并显示了基质特征与乳腺癌存活率之间的关联。 Lee等人还证明,前列腺癌中肿瘤相邻良性组织的计算分析可以揭示病理学家通常忽略但与无进展生存相关的信息。最近,Lu等人表明,描述核形状和核定向的特征与口腔癌和早期雌激素受体阳性乳腺癌的存活率密切相关。在许多情况下,免疫组织化学染色的可用性,使用抗体靶向图像中的特定蛋白质并标记特定的细胞和组织类型,避免了通过形态学检测细胞和组织的需要,从而能够生成复杂的数据,而无需使用DL工具。然而,在免疫肿瘤学的情况下,ML允许高通量生成描述数千个细胞的空间关系的特征,这对于病理学家来说是不可行的任务。通过DL方法对个体细胞和组织检测的改进允许非常精确地测量肿瘤微环境,因此现在可以大规模测量描述细胞和组织结构之间的空间关系的异质特征(图5)。
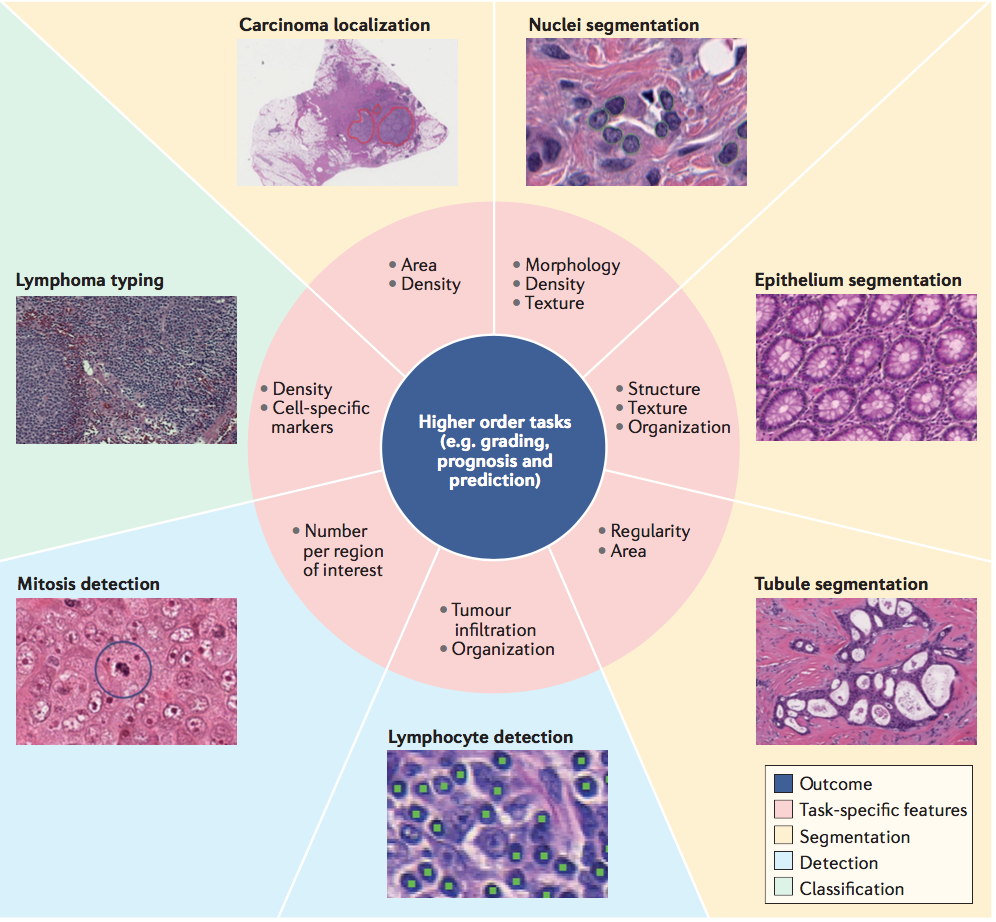
图5 | 机器学习应用程序的计算病理学任务。 深度学习框架可以使用图像分割(黄色背景)、检测特定特征(蓝色背景)、检测用于分类的一组特征(绿色背景)替代几种基本病理图像识别任务(如细胞核,上皮细胞或小管的分割,淋巴细胞检测,有丝分裂检测或肿瘤分类)中的传统手工特征, 。 识别基于粉红色区域中显示的任务特定特征,并且可以导致更准确的预后或疾病预测。
在Mani等人的一项研究中,利用淋巴细胞的几种标记物来理解这些人群在乳腺癌中的异质性。 Giraldo等人研究了细胞 - 细胞相互作用,并表明,利用细胞密度和PD1 +和CD8 +细胞的相对位置,他们可以鉴定出对Pembrolizumab有反应的Merkel细胞癌患者。 这些类型的实验的折衷是它们使用大量组织,通常每个染色需要额外的载玻片; 然而,可以检查数百或数千个特征,并且随着每个染色使用,可能的细胞 - 细胞相互作用的数量增加。 在这种情况下,特征选择和ML方法的组合用于确定可预测治疗响应的组合。
CNN在病理图像中的应用效果很好,因为有大量可行的像素可用于单次活检或切除的训练。 如果有足够精心设计的示例,可以设计DL算法来自动学习各种分类任务的功能。 例如,多尺度卷积神经网络(M-CNN)被用于监督学习方法中,用于在单个步骤中对高内容细胞图像进行表型分类,而不是几个独立的定制步骤。 仅使用来自图像的像素强度值将这些图像转换成表型,该方法导致对多种浓度的化合物处理的效果进行总体上更准确的分类。 许多图像分析挑战已经成功地使用DL方法来识别癌症肿瘤,肾小管,有丝分裂活动和乳腺癌和肺癌中的淋巴细胞内的区域。
除了病理图像之外,DL还可以促进其他信息形式的整合。 DL还可用于加速磁共振成像(MRI)数据采集或减少计算机断层扫描(CT)成像所需的辐射剂量。 利用包括时间和空间分辨率以及高信噪比的改进的成像质量,图像分析的性能可以相应地改进诸如图像量化,异常组织检测,患者分层和疾病诊断或预测的应用。 最近的另一项研究证明了使用初始DL框架来预测来自肺肿瘤的H&E染色图像的某些突变基因的存在的能力。
然而,尽管DL在许多特定图像分析任务中继续表现优异,但实际上,DL和传统图像分析算法的组合应用于大多数问题集中。 这样做有几个原因。 首先,虽然DL在非常具体的问题(例如肾小球的检测)中显示出其匹配或优于人类的能力,但它仍然不是一个很好的通用图像分析工具。 由于缺乏灵活性,开发时间很长。 特定分类任务的专家标签总体上也很少,因为这些标签生成起来很昂贵。 减轻这种情况的方法包括使用免疫组织化学染色为病理学家提供注释具有挑战性的样本的额外信息,以及为广泛使用病例(癌细胞与正常细胞)提高精心策划的专家注释的可用性的努力, 正在进行的社区任务。
另一个挑战是transparency问题。 已知DL方法的黑盒方法。 决定分类任务背后的基本原理尚不清楚。 对于药物开发,理解机制很重要,并且具有可解释的输出可用于不仅发现新的潜在药物靶标而且还可用于预测治疗反应的新潜在生物标志物。 为了增加对可解释性的信任,需要生成更多手工制作的功能。
另一个挑战是临床试验中需要大量样本以直接应用DL来推断治疗反应。 DL通常需要数万甚至数十万个例子才能学习,而临床试验通常不会产生足够的例子。 在某些情况下,有可能在临床试验中组合数据,但可能存在偏差,使得结果更难以解释。
成功整合DL和传统图像分析工作流程的例子包括Saltz等人和Corredor等人的工作,其中CNN用于检测H&E染色组织中的淋巴细胞,并且随后提取基于图形的特征以预测疾病反应。 这可能是DL在不久的将来的共同作用,因为其检测细胞和组织的卓越能力可以取代传统的分割和核检测算法,并且可以应用随后的可解释特征来给这些特征提供空间背景。
三、展望
DL方法和DL的最新发展为提高药物发现和开发渠道的效率提供了许多机会。 因此,我们预计未来几年将会有越来越多的应用程序针对整个行业明确定义的问题。 随着可用数据变得“更大”,至少在更彻底地覆盖整个数据空间的相关可变性的意义上,随着计算机变得越来越强大,ML算法将系统地生成改进的输出,并且新的,有趣的应用是 预计会跟进。 这已经在前面的章节中清楚地举例说明,其中我们已经描述了用于靶标识别和验证,药物设计和开发,生物标记物鉴定以及临床中疾病诊断和治疗预后的病理学的一些ML应用。
这些方法也在医疗保健环境中应用,当与药物发现相结合时,可以导致个性化医疗的显着进步。 ML还应用于电子健康记录和现实证据,以改善临床试验结果并优化临床试验资格评估过程。例如,最近的一项研究表明,DNN是一种高度竞争性的方法,用于从电子病历中自动提取有用信息,用于疾病诊断和分类。一些研究表明,电子健康记录中的ML模型在预测预后方面可以胜过传统模型。 ML还可以应用于现在来自传感器和可穿戴设备的数据,以了解疾病并开发治疗方法,特别是在神经科学方面。 Gkotsis等人应用DL方法来描述非结构化社交媒体数据的心理健康状况,这对于传统的ML方法来说是一项艰巨的任务。
如图1所示,ML方法开始在制药公司的发现和开发管道的各个步骤中普遍使用。 ML方法的这种普遍实现具有一些但是重要的已知问题。
-
深度训练的神经网络的典型问题是缺乏可解释性,即难以从训练的神经网络获得关于其如何到达结果的合适解释。如果该系统用于诊断诸如黑色素瘤的疾病,例如,基于医学图像,这种缺乏可解释性可能会阻碍科学家,监管机构,医生和患者,即使在神经网络比人类专家表现更好的情况下。患者是否比人类专家更信任ML诊断?尽管不那么引人注目,但药物设计中可能会出现类似的情况。一家制药公司是否会信任一个神经网络来选择一个小分子纳入其投资组合并投资进入诊所,而没有明确解释为什么神经网络选择了这个分子?此外,如果化合物已经通过计算机算法设计,则可能存在具有发明人的专利申请问题。无论如何,ML结果必须被视为只有假设或有趣的起点,然后由研究人员在研究中进一步发展。 验证ML结果的补充实验将有助于建立对方法和输出的信任,但监管机构尚未澄清他们对ML临床使用缺乏可解释性的看法。然而,即使超出了信任问题,这些方法缺乏可解释性,使得在新的看不见的数据集意外失败时对这些方法进行故障排除变得更加困难。
-
神经网络的另一个重要问题是重复性,这是因为ML输出高度依赖于网络参数的初始值或权重,甚至是训练样例呈现给网络的顺序,因为所有这些通常都是随机选择的。 。网络是否总是使用与输入相同的表达数据选择相同的疾病目标? ML方法提出的药物结构总是一样的吗?这种重复性的缺乏对于生物标志物鉴定尤其成问题,如在不同工具基于分子表达特征产生乳腺癌的不同预后生物标志物的情况中所见。不同的ML方法可以产生不同结果的事实将增加这些方法的大规模采用的不确定性。已经提出了解决可解释性和可重复性问题的一些解决方案。这些通常集中在使用更复杂或更耗时的算法或平均来自多个网络模型的结果,但这可能被视为仅在一系列现有结果中添加一个结果。
-
另一个需要考虑的重点是大量提供高质量,准确和精选的数据,以训练和开发ML模型。所需数量和准确度的要求取决于数据类型的复杂性和要解决的问题。因此,生成这些数据集可能很昂贵。使用适当数据标准并拥有必要的操作和开放数据框架的制药公司和学术机构的竞争前联盟可能是满足这些数据需求的解决方案的一部分。在药物发现过程中使用的许多数据类型远非全面。例如,蛋白质的所有折叠和结构的知识不完整,并且数据空间的覆盖范围同样不完整。因此,即使取得了很大进展,预测这些结构的应用还不如其他领域那么好。这同样适用于预测整个化学空间未知的小分子合成中涉及的反应。
-
数据管理是提供可重用且值得信赖的数据的关键,并且在所需的时间和技能方面可能是昂贵的。 生物管理(Biological curation) - 从科学文献中提取生物信息并将其整合到数据库中 - 介于艺术和科学之间,需要结合计算技能和深入的生物学和领域专业知识。 开发共享数据资源和元数据(标签)的协作努力可以是使公共领域中的高质量数据更加可用的方式。 这还包括来自成功和失败的药物发现计划的元数据,其可以实现预测方法和确定可以减少药物开发中的消耗的因素。 还需要更多的竞争前协作来汇总和生成研究化合物的企业生物活性数据集的大量数据资源以及历史临床试验数据。
-
ML模型应用中的另一个限制是用于预测替代范例。 因为ML的整个前提依赖于使用训练数据来生成合适的模型,所以ML模型只能在已知的训练数据框架内进行预测。 例如,在药物化学中,具有替代作用机制的化合物的设计,例如大环化合物,蛋白质 - 蛋白质相互作用抑制剂或PROTAC,可能仅可以用传统方法进行。
除了数据和模型之外,还需要加速对理解制药科学以及计算机科学,计算统计和统计ML以及熟练使用这些方法的研究人员的培训。 像DREAM Challenges(参见相关链接)这样的竞赛,表明团队组成是表现的一个因素,也可以吸引人才和推进方法开发。 但是,应用程序需要在临床环境中取得成功,以激励大型制药和技术公司的进一步投资。
ML算法(包括DL方法)使AI能够在行业环境和日常生活中得到应用。 ML方法在药物发现和保健的所有领域的影响已经被感受到,特别是在组学和成像数据的分析中。 ML算法在语音识别,NLP,计算机视觉和其他应用中也是成功的。 例如,支持因特网的智能助理现在很普遍,并且可以以语音和图像或视频的形式传输与健康相关的信息。 应用于从这种互联网技术融合中收集的数据的ML方法,加上生物数据,有可能显着提高此类算法的预测能力,并有助于医疗决策制定治疗效益,临床生物标志物和治疗副作用。
参考资料
- 11 April 2019. nature. Applications of machine learning in drug discovery and development. https://doi.org/10.1038/s41573-019-0024-5
