【5.4.1】先导化合物性质设计优化
一、药物的基本性质
1.1 药物的基本性质
有效性
- 与靶点相互作用 (基于结构的药物设计)
- 到达作用部位且达到有效浓度 (合理的药动学性质)
安全性
- 与非靶点作用
- 分布、结合、代谢 (药动学性质问题)
1.2 药代动力学(PK)
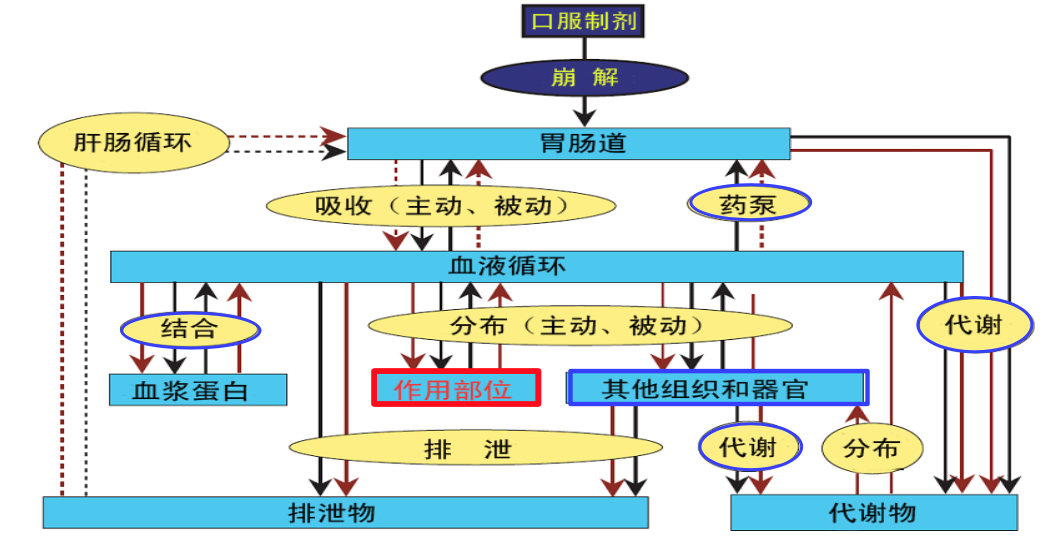
1.3 新药开发失败的主要因素
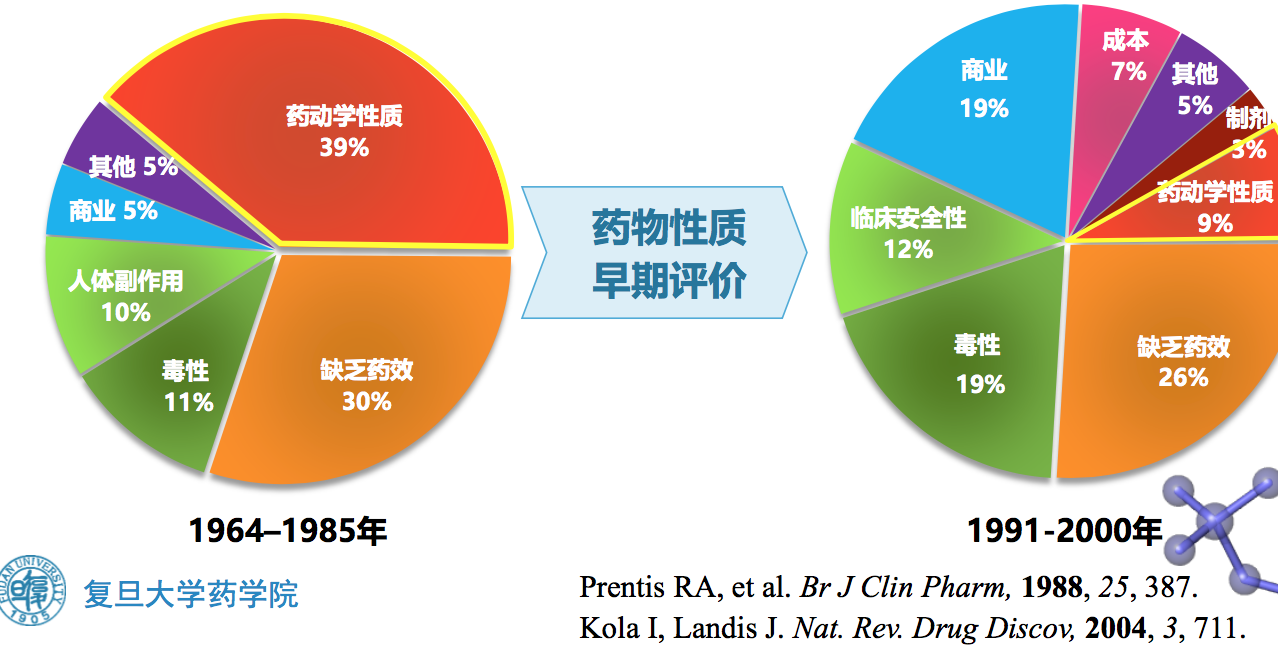
1.4 研发早期性质评价
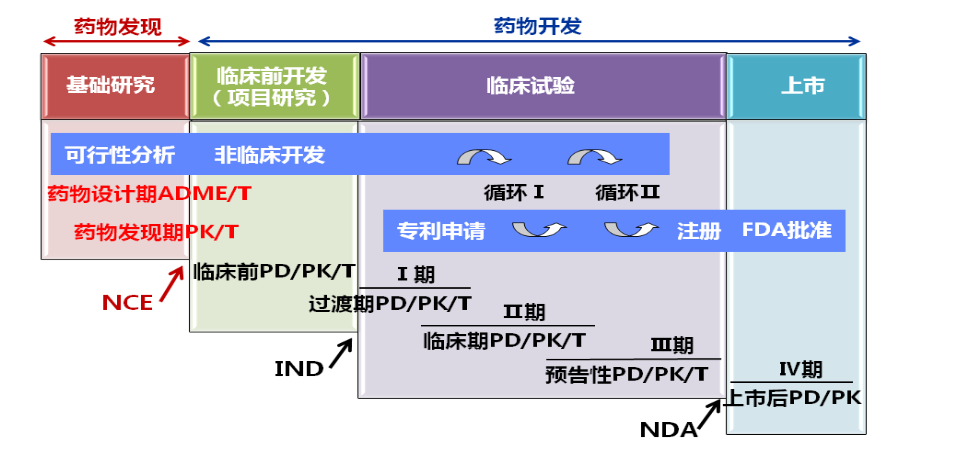
- 在可行性分析阶段,以适宜的药动学特征作为先导化合物优化的目标
- 在非临床开发阶段,以成药性作为候选药物评价的指标
1.5 基于性质的药物设计
-
基于性质的药物设计(Property-Based Drug Design, PBDD): 针对先导化合物或候选药物的结构进行药代动力学性质的设计与优化 ,以实现先导化合物或候选药物的良好口服吸收(A)、定向分布(D)、 可控代谢(M)、优化消除(E)或降低毒性和不良反应(Tox)。
-
基于性质的药物设计优化的最终目标:获得具有合理药代动力学性质 的候选药物。
二、构动关系和药动基团(SPR and Kinetophore)
- 构动关系 (Structure-Pharmacokinetics Relationship, SPR): 化合物的结构特征和理化性质与其ADME特征和药动学性质之间 的关系。
- 结构-性质关系 (Structure-Property Relationship, SPR)
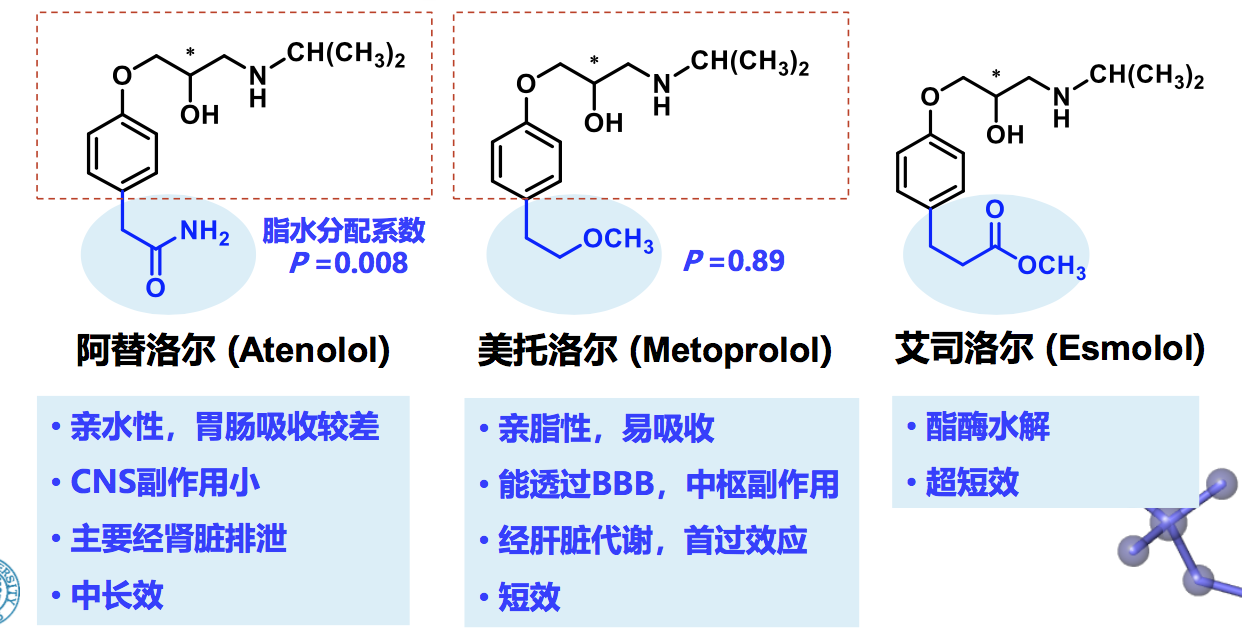
- 药动基团 (Kinetophore):化合物分子中影响ADME特征和药动 学性质的结构部分或官能团。
药动基团:β1受体拮抗剂
三、药物宏观性质与药动学性质的关系
3.1 建立药动学性质与药物宏观性质的关联

3.2 药物的宏观性质与PK性质
- 分子量:反映分子大小的参数,与透膜吸收和代谢有关
- 水溶性:口服吸收的前提,氢键可增加化合物的水溶性
- 亲脂性:亲脂性有利于跨膜被动扩散
- 离解性:与溶解性和透膜性密切相关
- 柔性:反映分子形状的重要参数
- 极性表面积:比氢键更能反映分子极性的真实情况
药物的宏观性质之一、分子量
- 分子量是选取先导物和临床候选药物的重要因素,对于提高新药研制 的成功率有重要意义
- 分子量大的化合物,功能基团多,增加了与受体结合的机会和强度
- 分子量过大影响化合物的水溶性,且被动扩散过程中难以渗透生物膜 上脂肪链有序紧密排列的磷脂双分子层,不利于透膜和吸收
- 分子量大的化合物可能含有易被代谢的基团和毒性结构,不适宜作为 先导物
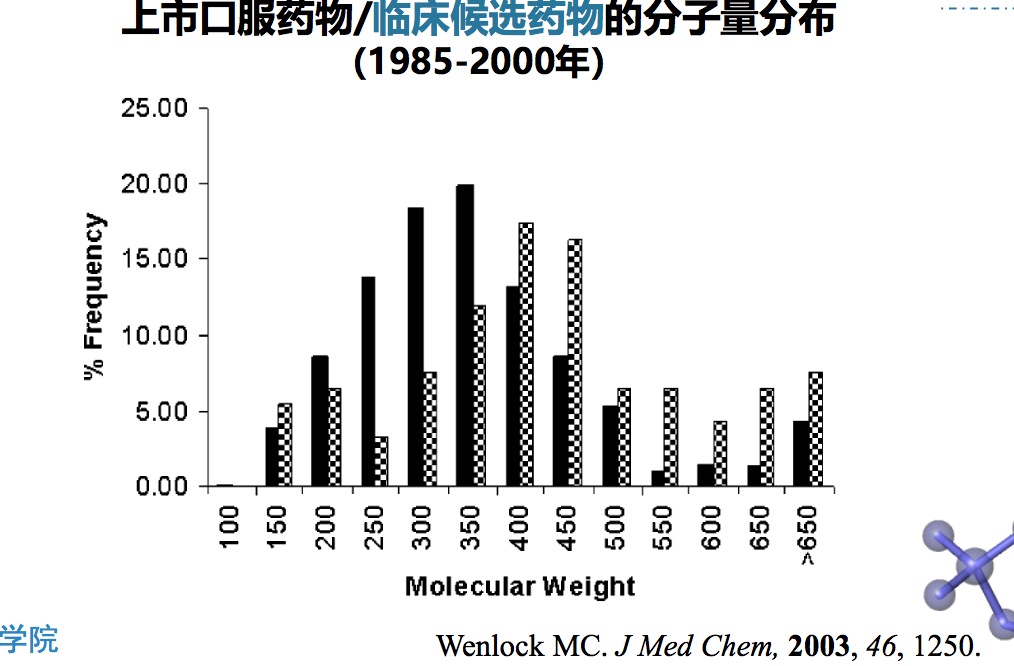
药物的宏观性质之二、水溶性
- 水溶性是口服吸收的前提条件,是穿透细胞膜的必要条件。
- 氢键可增加化合物的水溶性。
- 化合物只有在氢键破坏后才能渗透磷脂双分子层,因此,氢键数过多 不利于从水相经被动扩散分配到磷脂层。
- 水溶性影响候选药物在体内外的活性筛选质量
- 水溶性影响制剂的选择和质量

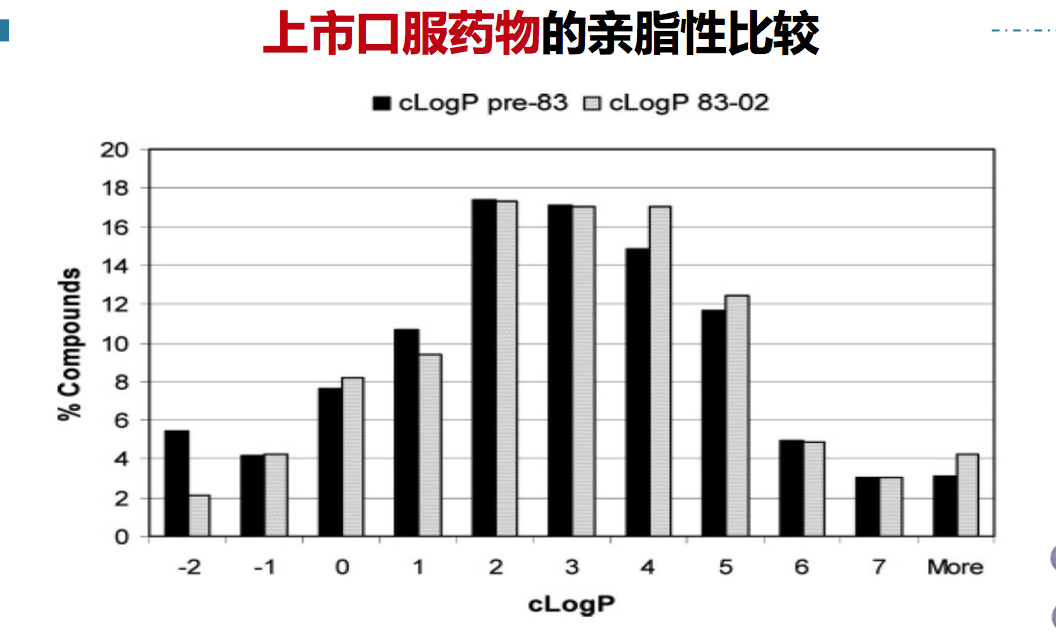
药物的宏观性质之三、亲脂性
适宜的亲脂性有多层次的贡献:
- 药效:亲脂性基团或片断参与同靶标的结合;
- 药代动力学:整体药物的亲脂性可影响透膜(穿透细胞膜磷脂层); 与血浆蛋白结合;组织分布(脂肪组织蓄积);穿越血脑屏障的能力; 代谢稳定性;
- 生物药剂学:影响药物分子在剂型中的溶出度、分散度以及制剂的 稳定性。
- LogP 和LogD7.4
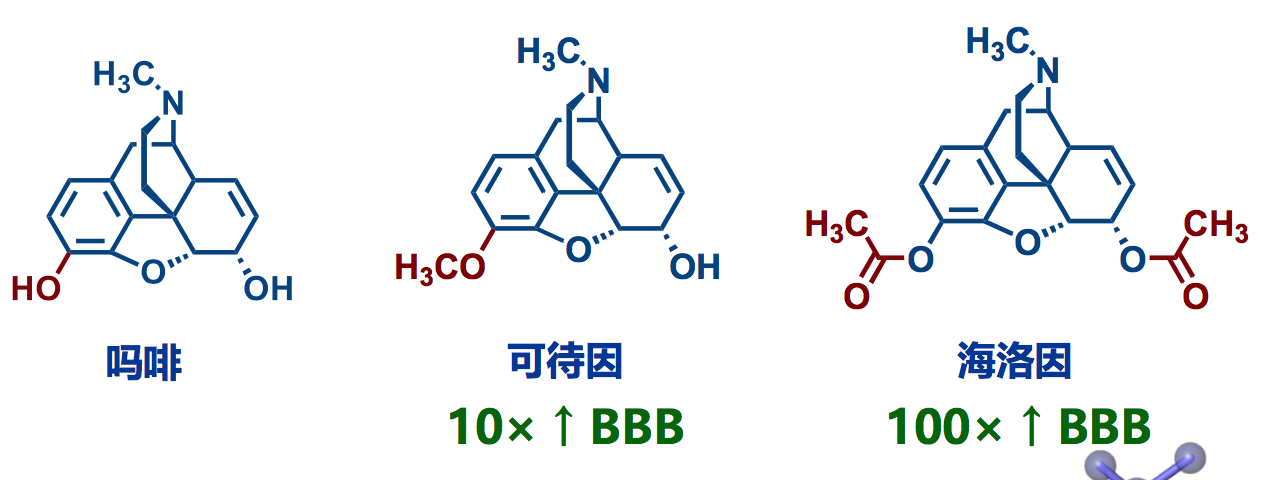
增加亲脂性,提高透脑率
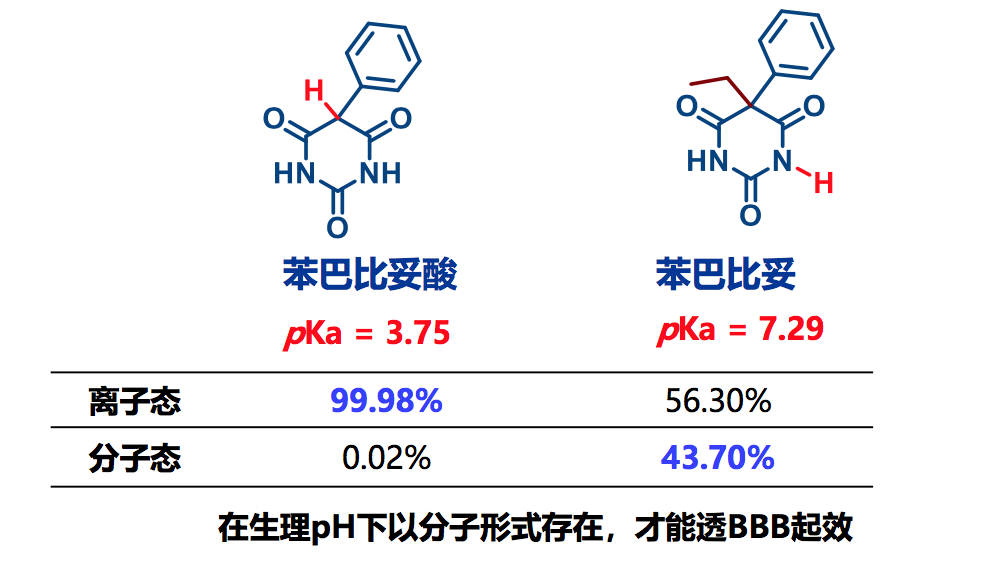
药物的宏观性质之四、离解性
- 离解性是化合物在介质中携带电荷的能力,与溶解性和过膜性 密切相关;
- 增加化合物的离解性增加了极性,提高水溶解度;
- 呈离解态的物质不利于过膜:强酸或季铵盐不利于过膜;碱性 过强也难以穿越BBB
- 可离解药物在不同pH环境下离解态和中性分子比例不同
- pKa
pKa对透膜性的影响
药物的宏观性质之五、柔性
- 分子柔性是反映分子形状的重要参数,在优化改造中要权衡利弊 * 增加柔性有利于分子与不同靶点发生结合。
- 分子柔性过高,不利于分散在水性环境中,也不利于透膜。
- 分子柔性参数通常用可旋转键数目(RB)表示。
可旋转键(Rotatable Bonds,RB)
- 可旋转键:与非末端的非氢原子连接的非环单键
- 酰胺C-N单键、磺酰胺S-N单键不可旋转

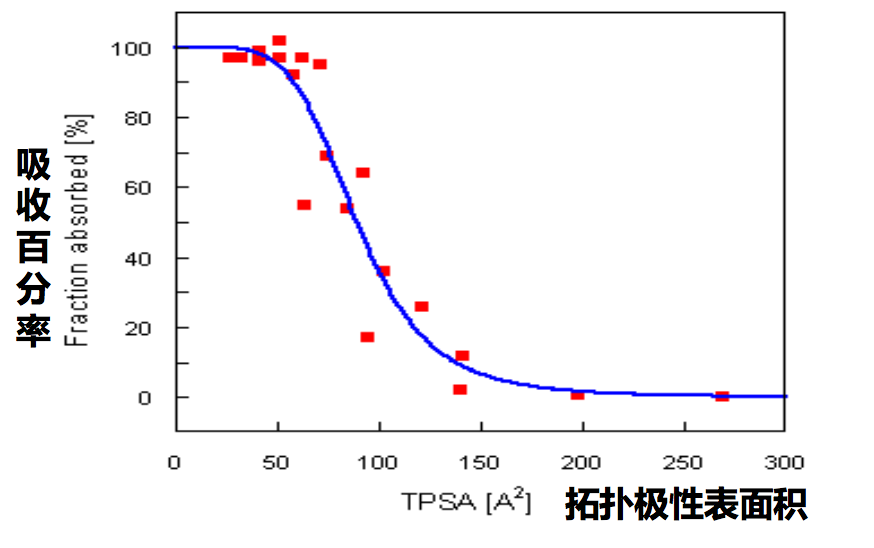
药物的宏观性质之六、极性表面积
- 极性表面积(Polar Surface Area, PSA)是分子中极性原子表面的 总和;比氢键更能反映分子极性的真实情况。
- 根据分子结构计算获得的极性表面积也称为拓扑极性表面积(tPSA)
- PSA越大,分子越难以过膜,吸收性越差
- 口服非CNS药物:PSA ≤ 140Å2
- CNS药物:PSA ≤ 90Å2
极性表面积与吸收性的关系
四、类药性与经验规则
4.1 类药性(Drug-likeness)
- 类药性是理想化药物应具有的良好性质,是药物所表现出来的 结构特征(如氢键供/受体、环结构、可旋转键数目等)和理化 性质(如相对分子质量、脂水分配系数等)在体内的综合反映 (ADME/T性质)。
- 类药性是对理想药物结构的要求
- 类先导性(Lead-likeness)是对先导化合物结构的要求
4.2 Lipinski规则——五倍率 (Rule of Five, Ro5)
- 相对分子质量 MW ≤ 500
- 计算脂水分配系数 CLogP ≤ 5
- 氢键供体数 (NH+OH) ≤ 5
- 氢键受体数 (N+O) ≤ 10
当不符合上述两项及以上规则时,化合物可能会出现 口服不吸收及透生物膜能力差的问题。
五倍率的适用性
- 仅适用于被动转运的情况
- 对抗生素、抗真菌药、维生素及强心苷等具有主动转运特征 的药物不适用
- 五倍率成为药物发现和虚拟筛选的标准环节
4.3 Veber规则
与大鼠口服生物利用度相关的规则——五倍率的补充:
- 可旋转键数目(rotatable bonds) RB ≤ 10 或分子中环的数目 rings ≤ 4
- 极性表面积(polar surface area) PSA ≤140Å2 或氢键总数(供体和受体)≤ 12
可旋转键(rotatable bonds, RB):
- 可旋转键是指与非末端的非氢原子连接的非环单键
- 酰胺C-N单键、磺酰胺S-N单键是不可旋转的
- 可旋转键数与分子柔性相关,影响药物吸收

其他规则
Clark’s BBB rules:
• N+O≤6
• PSA ≤ 90Å2
• MW≤450
• LogD=1-3
• CLogP-(N+O)>0
Rule of 3 (Lead-like):
• N+O≤3
• NH+OH ≤ 3
• MW≤300
• CLogP ≤ 3
• RB≤3
五、成药性的概念及其体内外评价方法
5.1 类药性与成药性的概念
-
类药性(Druglikeness)代表了理想化药物所具有的特点, 是药物所表现 出来的结构特征(如环结构、可选转键数目等)和理化性质(如相对分子 质量、脂水分配系数等)在体内的综合反映(ADME/T性质)。
-
类药性是对理想药物结构的要求;类先导性是对先导化合物结构的要求。
-
成药性 (Developability) 是药物除药理活性以外,与药代动力学和安全性 (PK/T)相关的所有其他性质, 是化合物能够进入临床I 期试验的特征指标。
-
成药性是对药物或候选药物(Candidate)属性的要求。
——郭宗儒. 药物分子设计的策略: 药理活性与成药性. 药学学报 2010, 45 (5): 539−547
5.2 成药性 (Developability)
决定候选药物是否能够转化为临床应用药物的性质

5.3 成药性测定方法
5.4 成药性优化
- 成药性是药物所表现出来的结构特征和理化性质在体内的综合 反映(包括药动学性质和毒性)。
- 成药性的表观性质是药代动力学和安全性,但归根结底是由分 子的结构特征所决定。
- 成药性优化的根本,实质上是化学结构的优化。
六、CYP450抑制作用 与药物毒性
6.1 细胞色素P450家族同工酶
细胞色素氧化酶P450(Cytochrome P450 proteins,CYP450)
- 一组结构和功能相关的超家族基因编码的同工酶,由血红素蛋白、黄素蛋白 (NADPH-细胞色素C还原酶)及磷脂三部分组成。
- 主要存在于肝脏细胞平滑肌内质网内,也称肝微粒体混合功能氧化酶。
- 在还原状态下可与CO结合,在波长450nm处有特征强吸收峰而得名。
- 参与体内内源性物质(如脂肪酸、维生素、胆酸)和外源性物质(如药 物 )的代谢,主要负责化合物体内的氧化、还原和水解3种I相代谢反应, 使它们易于从体内排出。
P450酶系被认为已经存在了350万年,在细菌、真菌及动植物中都能发 现其存在。
6.2 细胞色素P450酶的命名
- 族:40%以上的同源性,族用阿拉伯数字表示,如CYP1、CYP2
- 亚族:55%以上的同源性,亚族用大写英文字母表示,如CYP1A、CYP2D
- 具体的酶:用阿拉伯数字区分,如CYP1A2、CYP2D6
6.3 特点与临床意义
CYP450酶的特点:
- 特异性低
- 活性有限
- 个体差异大
- 可被药物诱导或抑制
CYP450酶的临床意义:
- 底物竞争性抑制
- 非线性药代动力学(安全性小)
- 基因多态性,个体化治疗
- 药物代谢性相互作用
6.4 与药物代谢相关的CYP450酶
- 涉及药物代谢的主要为CYP1、CYP2、CYP3家族,约占肝 脏总CYP450酶的70%
- 7个重要的CYP450:CYP1A2、CYP2A6、CYP2C9、 CYP2C19、CYP2D6、CYP2E1和CYP3A4
- 承担了CYP450酶系90%的功能和作用,参与了70%的药物 代谢,肠道I相代谢70%由CYP3A4催化
6.5 细胞色素P450的抑制类型
- 竞争性抑制:两种药物都是同一个酶的底物时,会产生底物之 间的竞争,抑制彼此的代谢
- 机制基础抑制:也叫自杀性抑制,如大环内酯类经CYP3A4代 谢,代谢物可与P450分子中的血红蛋白中的亚铁形成亚硝基 烷烃复合物而失活
- 非选择性抑制:指药物对多个同工酶都有抑制作用,缺乏选择 性。如西咪替丁可以同时抑制CYP1A2、CYP2D6、CYP3A4
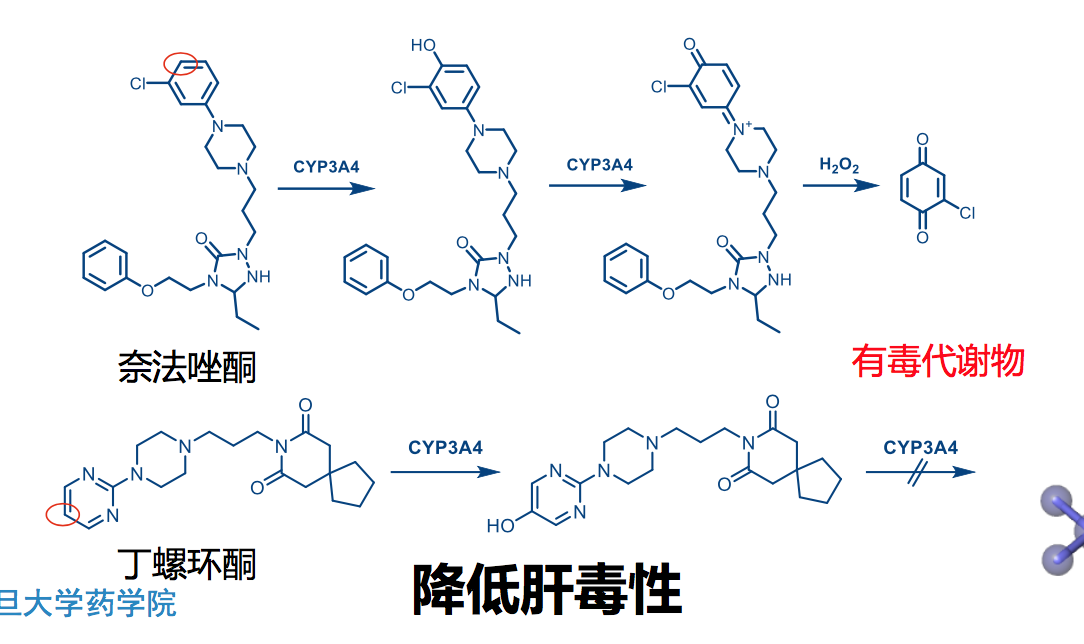
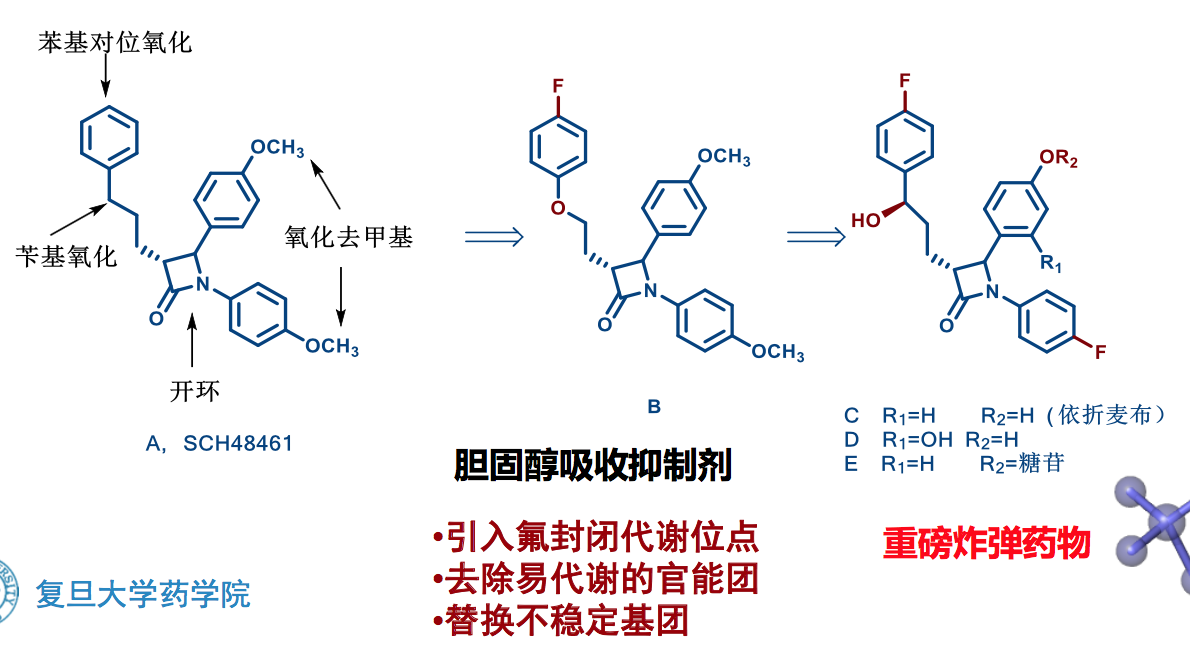
6.6 针对P450的药物改造实例
防止CYP3A4代谢的结构修饰

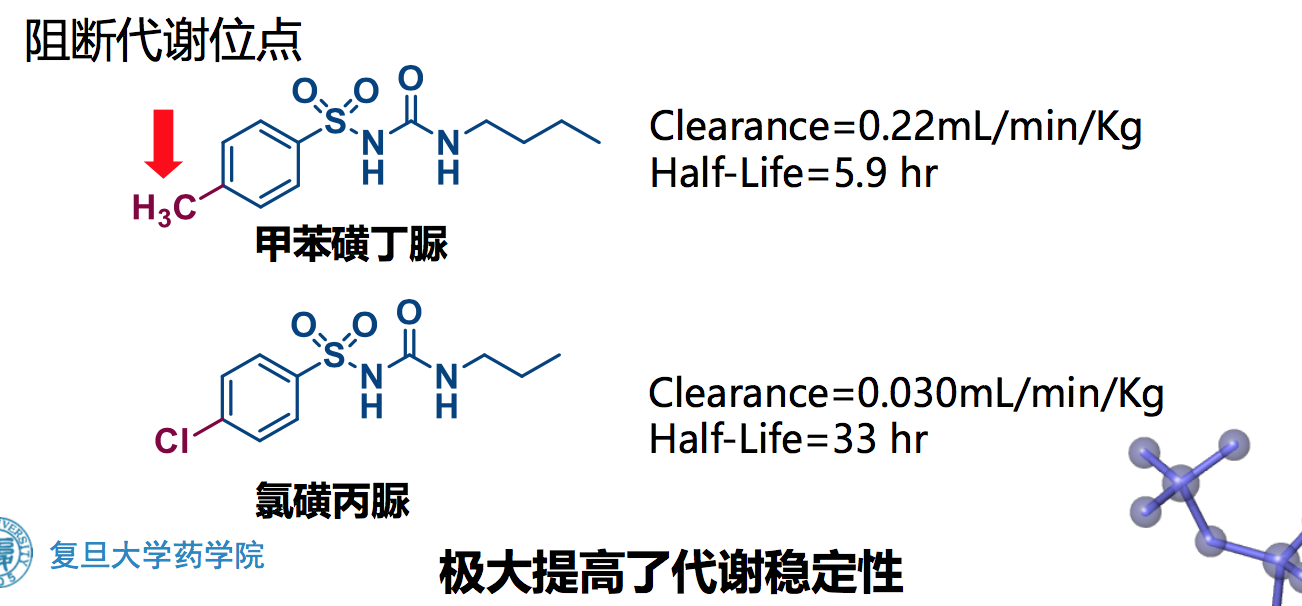
阻断代谢位点
七、hERG毒性及药物优 化策略
7.1 hERG简介
-
在心肌细胞中,hERG (human Ether-a-go-go Related Gene)编码钾通 道蛋白(Kv11.1),该蛋白介导一种延迟整流钾电流(IKr)。
-
抑制心肌细胞中的hERG会诱发获得性QT间期延长综合症(LQTS),导致严 重的心律失常。
-
目前,检测化合物对hERG钾通道的作用已是临床前评价化合物心脏安全 性的关键步骤,也是美国食品药品管理局(FDA)和欧洲药品评价局(EMEA) 等管理机构要求的新药报批必备资料,以此规避药物心脏毒性的风险。
7.2 药物心脏毒性的机制及应对
药物引起QT间歇延长的主要机制:
- 直接抑制hERG钾通道蛋白
- 阻碍hERG钾通道蛋白的转运
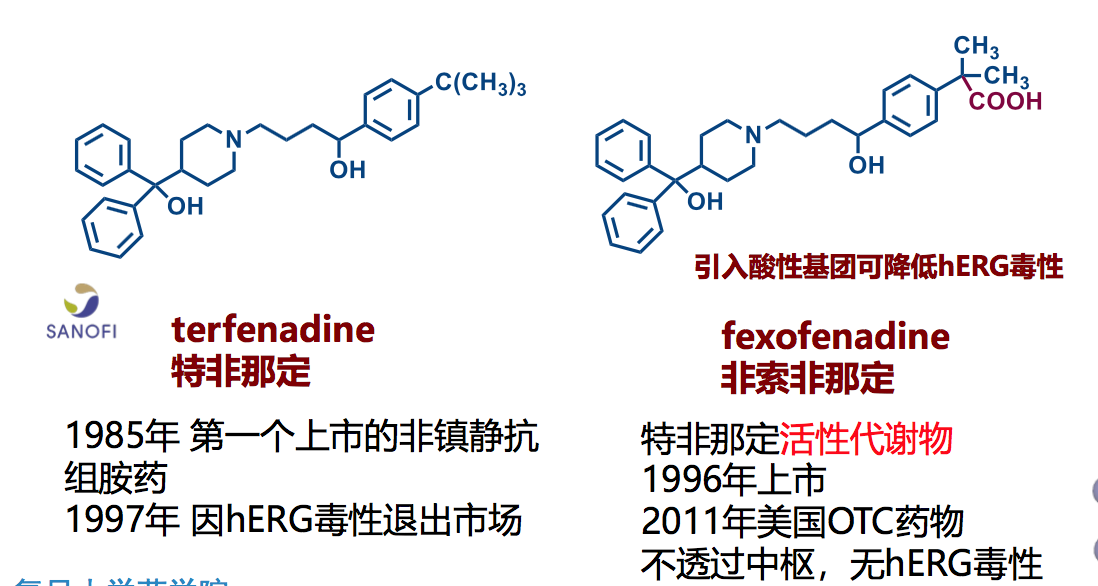
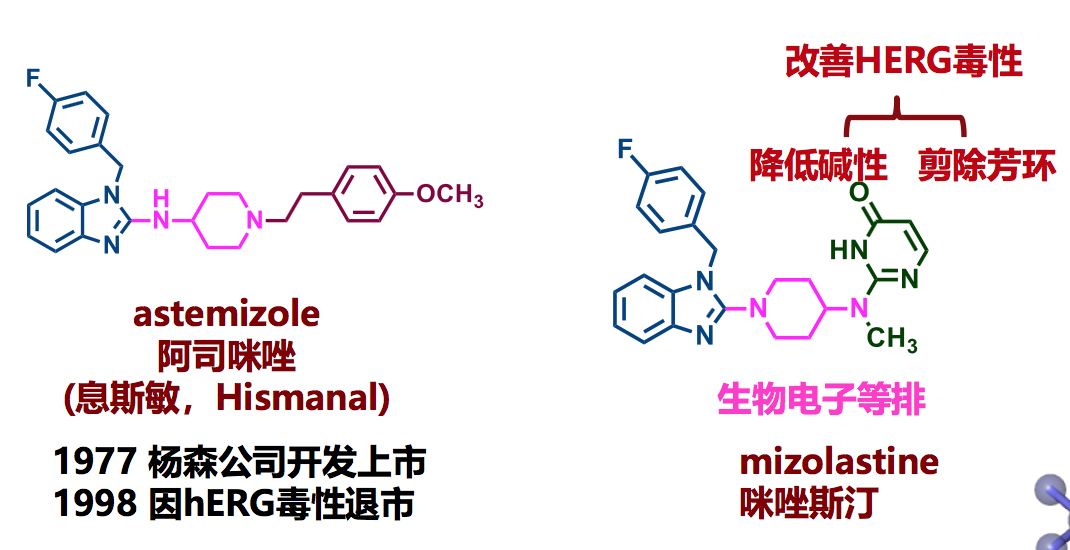
降低药物hERG毒性的主要策略:
- 降低脂溶性
- 降低碱性,增加酸性
- 构象限制
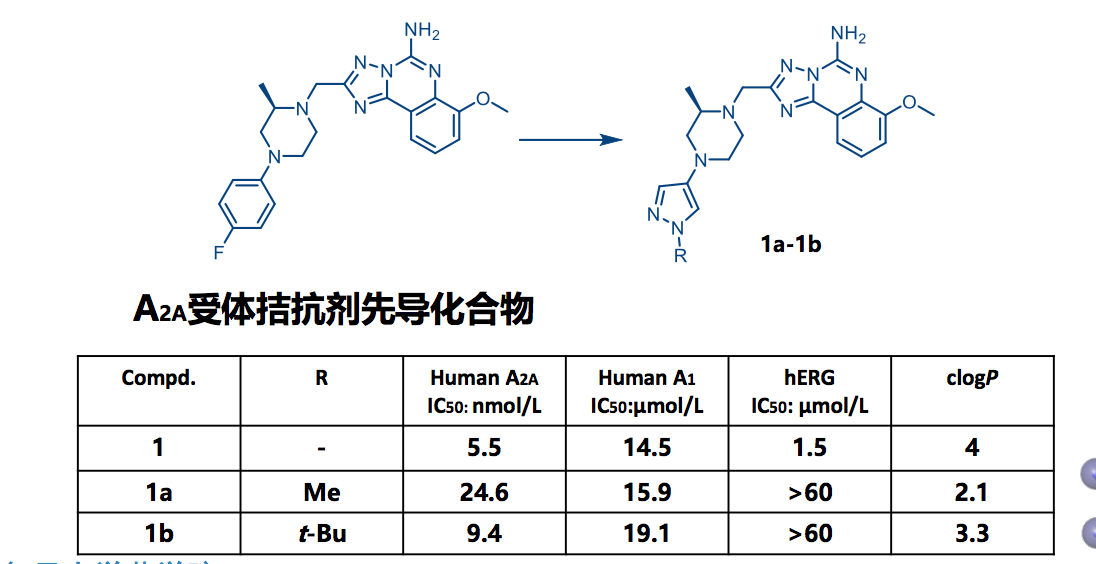
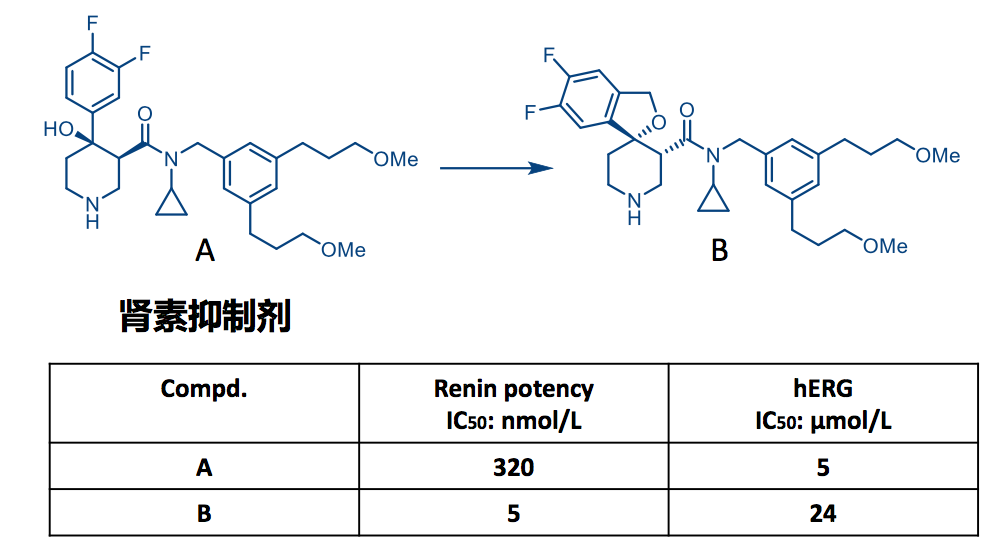
7.3 降低药物hERG毒性实例
参考资料
- 复旦大学 谢琼、周璐 老师的 《药物设计学》 课件
