【4.3.3】基于序列预测线性BCE--Bepipred-2.0
官网工具: http://tools.iedb.org/bcell/
The BepiPred-2.0 server predicts B-cell epitopes from a protein sequence, using a Random Forest algorithm trained on epitopes and non-epitope amino acids determined from crystal structures. A sequential prediction smoothing is performed afterwards. The residues with scores above the threshold (default value is 0.5) are predicted to be part of an epitope and colored in yellow on the graph (where Y-axes depicts residue scores and X-axes residue positions in the sequence) and marked with “E” in the output table. TheÊvaluesÊof the scores are not affected by the selected threshold. The table below shows the relationship between selected thresholds and the sensitivity/specificity of the prediction method.
Threshold Sensitivity Specificity
0 1 0
0.05 1 0
0.10 1 0
0.15 1 0
0.20 1 0.00019
0.25 0.99743 0.00419
0.30 0.98995 0.0276
0.35 0.97212 0.07036
0.40 0.93605 0.15606
0.45 0.82607 0.3307
0.50 0.58564 0.57158
0.55 0.29159 0.81655
0.60 0.09559 0.95116
0.65 0.01969 0.99272
0.70 0.00182 0.99954
0.75 0.99743 0.00419
0.80 0 1
0.85 0 1
0.90 0 1
0.95 0 1
1 0 1
抗体已成为许多生物技术和临床应用中必不可少的工具。它们通过高度特异性地识别其分子结构(抗原决定簇)的一部分来结合分子靶分子(抗原)。仅从抗原序列预测表位的能力是一项复杂的任务。尽管付出了巨大的努力,但在过去的十年中,表位预测方法的准确性取得了有限的进步,特别是对于仅依赖抗原序列的方法。在这里,我们介绍了BepiPred-2.0( http://www.cbs.dtu.dk/services/BepiPred/ ),这是一种根据抗原序列预测B细胞表位的网络服务器。 BepiPred-2.0基于随机森林算法,该算法在从抗体-抗原蛋白结构注释的表位上进行训练。发现这种新方法的性能优于其他可用于基于序列的表位预测的工具,这些工具既可用于从已解决的3D结构派生的表位数据,又可用于从IEDB数据库下载的大量线性表位。该方法以计算机友好和非专家用户友好且信息丰富的方式显示结果。我们相信BepiPred-2.0将成为生物信息学和免疫学界的宝贵工具。
一、前言
B细胞被认为是适应性免疫系统的核心组成部分,因为它们具有识别并长期抵御传染性病原体或癌细胞的能力。它们通过产生抗体,在B细胞表面上分泌或表达的蛋白质,以及通过以高度选择性的方式与一部分分子靶标(称为表位)结合而识别其分子靶标(称为抗原)的蛋白质来执行这些功能。在疫苗中采用了这种识别过程,以使用减毒和亚单位疫苗等不同方法对所需病原体提供长期保护。
B细胞表位可以分为两组。线性表位由抗原蛋白序列中的残基的线性延伸形成。相反,不连续的(构象性)表位是由抗原序列中相距很远的残基形成的,这些残基通过折叠而在空间上聚集在一起。即使大多数表位是构象的,大多数也包含一个或几个线性延伸(1)。
可靠的B细胞抗原决定簇预测工具在许多临床和生物技术应用(例如疫苗设计和治疗性抗体开发)中以及对于我们对免疫系统的一般理解中至关重要(2-4)。
已经开发了几种基于结构的工具,可在已知抗原结构时用于预测和分析表位(5-9)。但是,结构信息仅可用于非常小部分的抗原,并且在大多数情况下,只需要分析一级序列即可。这样的基于序列的预测器的准确性通常很差,并且在过去几年中几乎没有取得任何进步。在大多数情况下,当前方法的培训是基于经过实验验证可与抗体结合的肽(10-13),并且通常与预测工具的性能低下有关(4),这可能是由于起始数据的注释不清晰和嘈杂。
在这里,我们介绍BepiPred-2.0,这是一个用于基于序列的B细胞表位预测的Web服务器。与BepiPred-1.0不同,BepiPred-2.0仅在源自晶体结构的表位数据上进行训练,与其他可用工具相比,这被认为具有更高的质量,并且确实大大提高了预测能力(10,11)。
二、材料和方法
我们简要描述了用于训练BepiPred-2.0的数据集和方法,以及我们已经执行的验证。有关材料和方法的更多详细信息,请参见补充材料。
2.1 结构数据集
从蛋白质数据库(PDB)获得了由649种抗原-抗体晶体结构组成的数据集(14)。在每种复合物中,我们使用在别处开发的HMM模型识别抗体分子,对于每种抗体,我们将其抗原定义为所有非抗体蛋白链,这些蛋白链从其互补决定区(CDR)的半径4Å中具有至少一个原子原子(15)。我们去除了抗原序列与我们数据集中的任何其他序列> 70%相同的复合物,从而获得了160个结构。我们随机选择了2014年之后发布的五个结构作为最终评估数据集,并使用剩下的155个结构(分成五个相等大小的分区进行交叉验证)来创建我们的训练数据集。表位残基定义为任何抗体残基重原子半径4Å中的那些。同样,如果多个相同的抗原链与同一抗体结合,则表位定义为所有链上的表位残基的并集,从而得到3542个残基的阳性数据集。所有36 785个非表位均定义为阴性。在评估方法的性能时,会使用所有正负残基,但为了进行训练,通过随机抽样将负数据集缩减为与正数据集相同的大小(有关更多详细信息,请参见补充材料)。
2.2 训练随机森林预测模型
为了预测给定抗原残基是表位的一部分的可能性,使用5倍交叉验证方法对随机森林回归(RF)算法进行了训练。使用NetSurfP(19)预测的每个残基的计算体积(16),疏水性(17),极性(18)以及相对表面可及性(RSA)和二级结构(SS)对其进行编码。大小为9的窗口以残基本身为中心。另外,使用了通过将所有抗原残基的各个体积相加而获得的抗原总体积,总共有46个变量。然后对RF输出执行窗口9的滚动平均值,以获得最终的BepiPred-2.0预测。有关参数优化的更多详细信息,请参见补充文本以及补充图S1和S2。
2.3 评估测量
我们根据接受者操作曲线(AUC)下的面积,接受者操作曲线的前10%下的面积乘以10(AUC10%)归一化的阳性预测率(PPR)评估了每种抗原的性能。以及前60个预测的真实阳性率(TPR)(20)。
比较两个模型的性能时,根据它们在单个抗原上的性能计算成对的t检验。 95%的置信区间用于定义两个比较模型之间的显着差异。
2.4 线性表位数据集的评估
从免疫表位数据库(IEDB)下载了一组已知的线性肽,这些肽进行了免疫识别测试,被发现是表位(阳性测定结果)或非表位(阴性测定结果)(21)。短于5个氨基酸或大于25个氨基酸的肽被去除,因为B细胞表位很少在这些边界之外(1)。阳性数据集中仅包含在两个或多个单独实验中被确认为阳性的肽,在阴性数据集中仅包含在两个或多个单独实验中被视为阴性但从未在任何实验中观察为阳性的肽。这产生了11 834个阳性肽和18 722个阴性肽。每个肽都重新定位在其原始蛋白质序列上,并用于计算输出预测。该数据集可从BepiPred网页( http://www.cbs.dtu.dk/services/BepiPred/download.php )下载。
仅对阳性和阴性肽内的残基进行评估。在这种情况下,仅对合并的阳性和阴性残基而不是每个抗原序列计算AUC。
三、网页界面
为了使用BepiPred-2.0( http://www.cbs.dtu.dk/services/BepiPred/ ),用户只需要Fasta格式的目标蛋白质序列即可。所有预测都是即时进行的,并且在几秒钟到几分钟内,主要取决于输入数据的大小,用户将被重定向到结果页面。在此,预测的表位在输入蛋白质序列中指示。支持所有最常见的浏览器,例如Chrome,Firefox,Microsoft Edge和Safari,但是Internet Explorer上不提供某些图形功能。在以下段落中,将进一步详细描述输入页面和输出页面。可以在BepiPred-2.0说明/帮助页面上找到一些提示和技巧以及有关Web服务器的更详细说明。
3.1 输入页面
通过将它们粘贴到文本框中或作为单个文件上传,用户最多可以提交50种fasta格式的蛋白质序列。不支持核酸序列,蛋白质序列应大于10个氨基酸且小于6000。单击“ Example Antigens”按钮时,将提供示例序列。单击“提交”后,用户将被重定向到作业队列页面,该页面每20秒更新一次。预测完成后,用户将自动重定向到输出页面。 (可选)用户可以提供电子邮件地址,并且在作业完成时将通过电子邮件发送结果页面链接。
3.2 输出页面
BepiPred-2.0输出页面包含带有各种选项卡的导航栏。 “摘要”标签在水平和垂直可滚动表格中显示每个单独的序列结果。默认输出格式显示每个序列的BepiPred-2.0预测和表位分类。 BepiPred-2.0预测用于设置蛋白质序列的背景颜色。在蛋白质序列本身上方的“表位”行中,所有大于用户定义阈值(默认为0.5)的预测都标记为“ E”。使用“表位阈值”滑块,可以根据需要修改表位分类。通过按阈值滑块旁边的“?”按钮,将显示每个阈值的预期灵敏度和特异性图。将鼠标悬停在序列上会显示特定残基的预测值,将鼠标悬停在蛋白质名称上会从fasta标头中显示出蛋白质的描述。单击“高级输出已关闭”按钮将切换到高级可视化模式,其中添加了NetSurfP的结构预测,如图1所示。这使有经验的用户可以显示详细信息并更好地解释结果。 “日志”标签将显示计算和已发生的错误的日志,“帮助”标签包含提示和技巧以及输出页面的详细说明。您可以使用“下载”下拉标签以JSON或CSV格式下载预测,点击“所有下载”即可找到文件的简短说明。

四、结果
在这里,我们演示了Bepipred-2.0(由大量结构定义的B细胞表位构成的Web服务器)的功能,并展示了此更新方法在结构和线性表位验证数据集上如何显着优于BepiPred-1.0(10)。
4.1 交叉验证结果
我们使用5倍交叉验证方法来评估Bepipred-2.0方法的性能。该数据集由源自165个已解析结构的表位组成,其中没有两个抗原共享> 70%的序列相似性。在该结构表位测试集上,最终的RF模型实现了0.62的AUC和0.121的AUC10%。
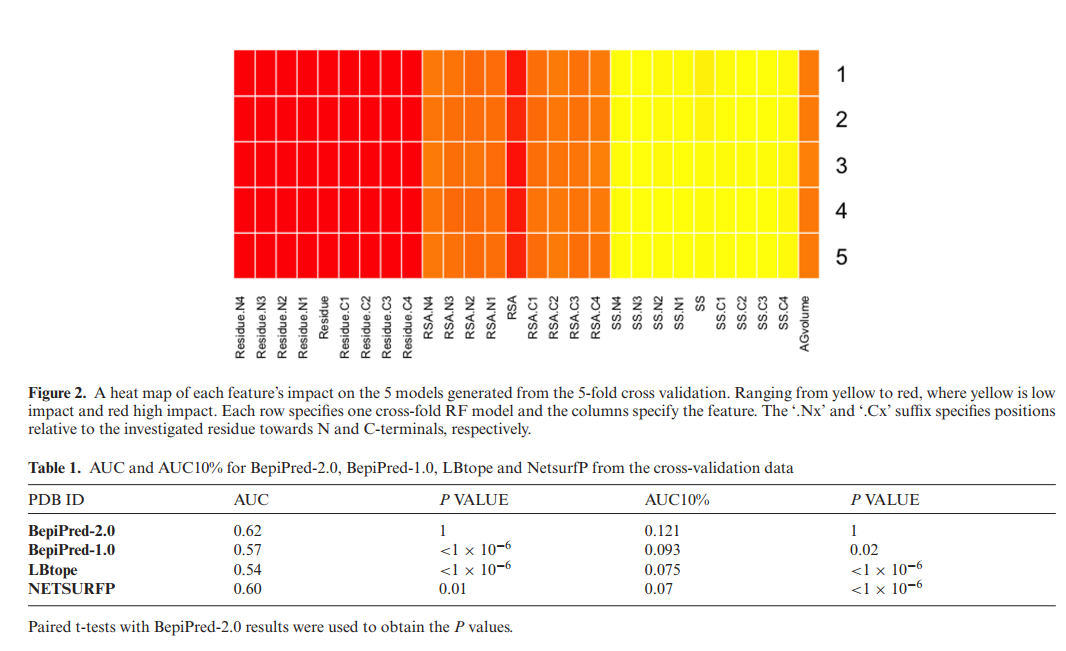
图2显示了基尼(Gini)重要性,描述了每个变量和交叉折叠分区(22)的每个功能的重要性。 5个RF模型的基尼重要性值高度一致,证实了所提出模型的鲁棒性。此外,该图表明,除残基类型外,预测的RSA是有助于我们方法预测能力的最重要特征之一。
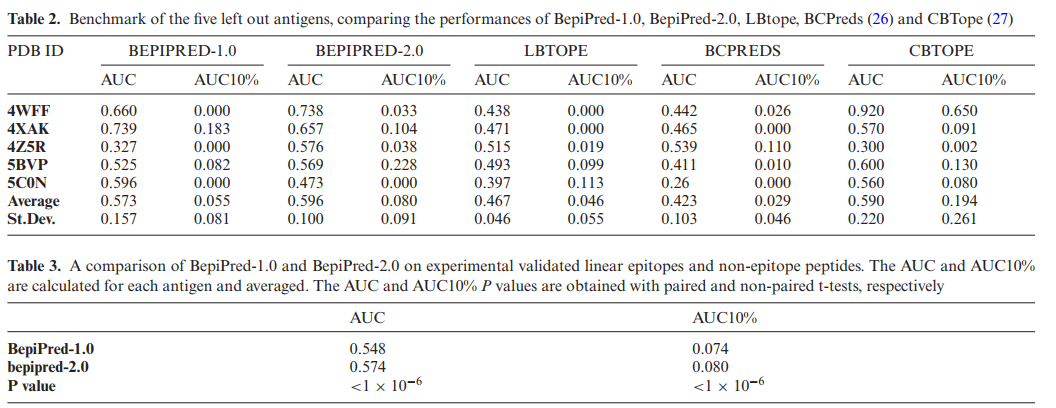
在此数据集上,BepiPred-2.0优于其他两种经过测试的方法,即BepiPred-1.0(10),LBtope(11),这两种方法都是用于线性表位预测的最常用方法。 同样,仅基于NetSurfP提供的RSA值,该方法的性能优于基线预测值。 表1显示了AUC,AUC10%和相应的p值。
在许多实际情况下,用户仅对分析得分最高的预测感兴趣,因为他们使用预测来确定一些候选者的实验测试优先级。 为了评估这种情况,计算了每种蛋白质的60个得分最高的残基的平均阳性预测值(PPV)和真实阳性率(TPR)。 从图3可以明显看出,与其他方法相比,BepiPred-2.0的PPV显着提高,TPR略有提高。
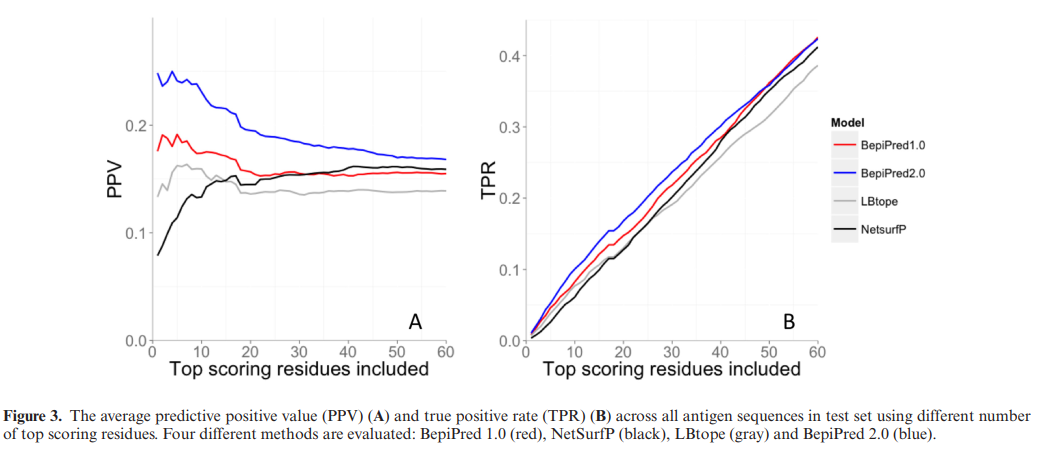
4.2 独立的结构表位基准
表2显示了评估数据集中的五个结构上BepiPred-2.0与其他方法相比的性能。 这些结果证实BepiPred-2.0具有比BepiPred-1.0和LBtope更高的性能。 特别是,由于AUC10%专注于最高预测,因此此措施的BepiPred-1.0和BepiPred-2.0之间的差距突显了2.0的特异性高于高得分残基的1.0。
4.4 线性表位预测能力的评估
为了进行公平的比较,在对BepiPred-1.0进行线性表位训练时,我们在由免疫表位数据库(IEDB,请参见 材料和方法)。 该基准测试的结果记录在表3中,表明该数据集上的BepiPred-2.0也优于BepiPred-1.0。
4.5 案例研究:溶菌酶表位区域
典型的抗体靶标(如溶菌酶)已与结合至不同区域的不同抗体形成复合物结晶,而大多数蛋白质仅由单一抗体结晶。以溶菌酶为例,我们可以看到当前在不同的已解析结构(1BVK,1C08、1MLC,4TSB)中存在四个独特的表位区域,如图4所示,其中溶菌酶根据BepiPred-2.0的预测进行了着色。重要的是要注意,如果我们一次仅对这四个表位之一评估我们的方法的性能,我们将获得0.593±0.171的平均AUC和0.127±0.211的平均AUC10%。另一方面,如果评估中包括所有表位区域,则BepiPred-2.0的AUC为0.713,AUC10%为0.304。该结果证实了较早的发现,即B细胞抗原决定簇预测相对较低的预测性能的可能主要原因是由于当前可用抗原决定簇基准数据的偏倚和不完整注释所致(9)。
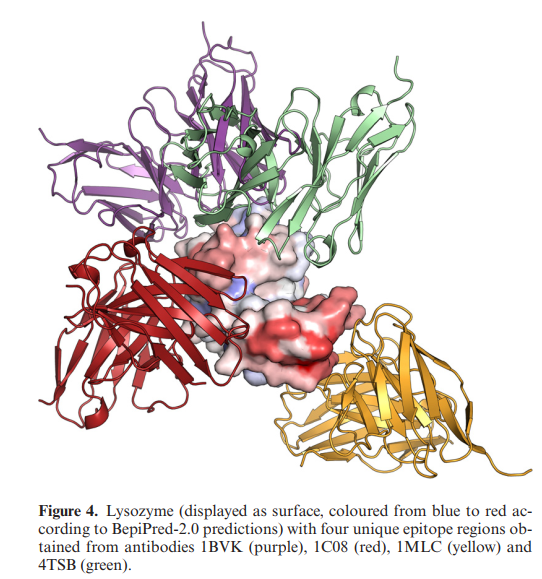
五、讨论
BepiPred-2.0 Web服务器提供了基于最新B细胞表位序列的预测。我们相信,直观的界面将帮助具有有限计算知识的研究人员充分利用和理解结果。此外,高级选项允许更多有经验的研究人员根据其他预测的结构特征进一步解释输出。
与以前的对线性肽段进行抗体识别测试的预测工具相比,使用晶体学得出的结构表位数据进行训练和评估可以显着提高性能。即使以与训练BepiPred-1.0相同的数据类型进行评估,BepiPred-2.0仍可显着提高性能。我们相信,这是一个重大发现,它将使其他人知道将来如何评估B细胞表位。此外,如溶菌酶实例所示,不同的抗体可以识别多个区域,这引发了如何正确定义表位的问题(23,24)。目前,我们和其他人(7,25)正在研究的一种可能的解决方案是开发可预测单个抗体或抗体文库特异的抗原上的表位区域的工具,从而提高预测的特异性。目前,抗原决定簇预测工具可以用作过滤器,以排除不太可能是进一步实验分析中的抗原决定簇的区域,但是随着这些工具的准确性和特异性的提高,我们相信它们将允许进行精确且有针对性的实验
参考资料
- Jespersen MC, Peters B, Nielsen M, Marcatili P. BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res 2017. 网址:https://academic.oup.com/nar/article/45/W1/W24/3787843
