【8.3】抗体的深度学习方法综述
在计算机视觉和自然语言处理等领域的成功推动下,深度学习最近通过协助细胞图像分类,发现基因组联系和推进药物发现而进入生物学领域。 在药物发现和蛋白质工程中,主要目标是设计一种分子,该分子将起治疗药物的作用。 通常,重点放在小分子上,但是已经开发出新的方法来将这些相同的深度学习原理应用于生物制品,例如抗体。 在这里,我们简要介绍了应用于抗体药物开发的深度学习背景,并深入解释了已提出的几种深度学习算法,这些算法可解决一般蛋白质设计,特别是抗体设计的各个方面。
一、前言
在本文中,我们概述了深度学习技术,这些技术已开始应用于抗体设计及其结果领域。 我们概述了抗体设计三个方面的当前挑战:
- 从序列进行结构建模;
- 预测蛋白质相互作用;
- 鉴定可能的结合位点。
然后,我们将探讨针对这些领域中的抗体设计而开发的深度学习技术和算法。 由于这三个挑战领域在一般的蛋白质空间中具有类似物,因此我们将在更广泛的范围内针对蛋白质之间的类似问题描述深度学习方法,并应理解这些方法可能适用于抗体工程的较窄域。 我们还将描述可用于帮助开发和比较该领域中的方法的数据集和基准。 我们将对这些方法进行比较,最后指出未来的方向。
单克隆抗体疗法已成为针对目标和适应症的药物开发越来越流行的方法,在这些方法中,基于小分子的方法已被证明不足。随着人们对焦点的日益关注,已经出现了许多改进和完善抗体开发流程的新方法。构建展示文库(噬菌体和酵母)的创新加快了候选发现的时间表,并减少了与治疗线索的下游开发相关的挑战。当前lead开发研究的一个重要目标是减少下游铅优化步骤的必要性,例如改善候选物的溶解度和免疫原性,以及减轻其他可开发性问题。其他研究通过添加其他功能域来创建双特异性抗体和Fc效应子抗体,以及通过探索具有独特特性的不同构建体(例如单链可变片段和骆驼科动物衍生的纳米抗体),扩展了潜在抗体治疗剂的作用机制。
尽管这些体外创新缩短了时间表,并改善了整个开发流程中的不同步骤,但围绕计算机模拟技术和抗体候选物的设计出现了一类新的创新。这些方法试图利用计算处理能力的进步来降低成本并提高潜在候选人的生成速度。电子计算机管道的优势将包括快速,廉价地扩展候选物的生成,开发针对具有挑战性的抗原的抗体的能力以及合理设计原则的应用。与传统的候选物生成方法(例如杂交瘤或噬菌体展示)相反,计算机模拟管道有望开发出更便宜,更快速的药物。但是,传统的计算机方法尚未完全兑现这些承诺。在这里,我们提出了基于深度学习的方法,在计算抗体设计的关键挑战方面,与传统方法相比,它们似乎表现出更大的成功。
二、抗体
抗体是作为对入侵病原体的免疫反应而产生的一种蛋白质。它们由四个链组成-两个重链和两个轻链。重链包括三个恒定域和一个可变域,而轻链仅具有一个恒定域和一个可变域。可变域包含抗体的结合表面或“互补位”。互补位主要由六个不同的可变环组成-轻链上三个(环L1,L2和L3),重链上三个三个(环H1,H2和H3)(图1)。该区域也称为互补决定区(CDR),可以使抗体以高特异性结合靶标[1]。该区域足够大,可以容纳许多独特的接触,这是实现如此高特异性的部分原因,尤其是与典型的小分子相比,后者可以容纳更少的接触并因此具有更多的副作用-导致脱靶互动。 CDR环之间的显着变化程度非常重要,因为抗体的多样性是使它们成为如此广泛的靶标的有效结合物的一部分[1]。

抗体的特异性和广泛适用性使它们成为医学研究的重点,而这反过来又引起了人们对以计算机或计算机方式进行抗体研究的关注。
为了计算分析抗体或预测其有效性,通常需要生成三维模型。由于传统的结构确定方法(例如X射线晶体学,核磁共振(NMR)和低温电子支气管镜检查(CryoEM))费力,费时且昂贵,因此出现了使用化学方法和现有蛋白质折叠数据生成结构预测的计算方法。几个小组已经能够准确地预测一组基准的抗体结构,但是H3 CDR环的建模仍然是一个重大挑战[2]。相对于其他CDR环,产生H3环的生物学过程是独特的。环的大部分由其自身的基因编码,与编码其余抗体序列的基因分开。其他CDR环的变异要小得多,甚至可以合理地分成典型的结构簇,而编码H3的基因在孤立的情况下会主动发生突变,然后在称为V(D)J重组的过程中与其余的基因序列重组,在这些loop之间会产生顺序性和构象性高变异性[3,4]。此过程引入的loop序列极其多样,因此几乎不可能与相似的loop同源。几乎没有足够的同源性数据来预测结构。这提出了巨大的挑战,这显然需要新方法。
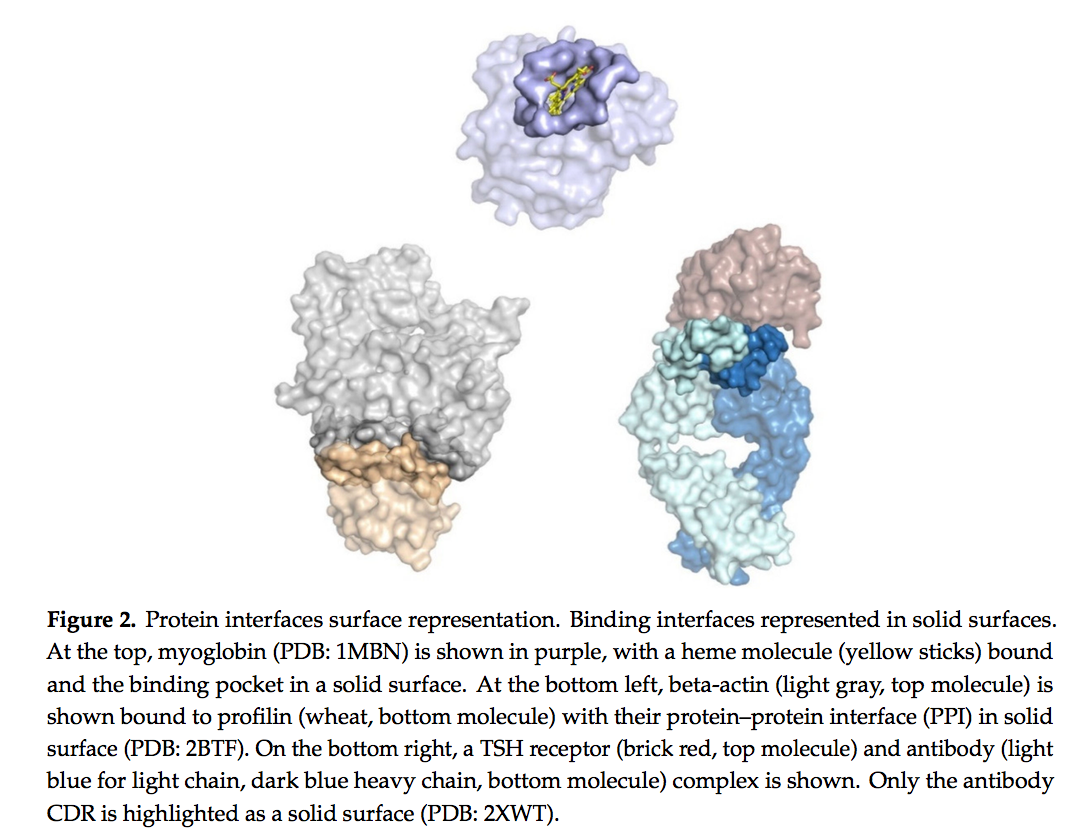
计算抗体开发中的另一个挑战是界面预测。通常,两种蛋白质之间的界面由形成紧密相互作用的几个保守良好的残基组成。其原因是通常两个相互作用的蛋白质将在许多世代中共同进化。如此长的共享历史无法使抗体与其抗原相互作用。抗体是“即时”产生的用于处理外来病原体的临时结合物。尽管抗体-抗原相互作用确实属于蛋白质-蛋白质相互作用(PPI)的总括,但越来越明显的是,抗体-抗原相互作用及其界面是不同的,其独特的性能降低了一般蛋白质相互作用预测抗体空间对蛋白质-蛋白质相互作用的适用性。。(图2)由于仅界面的抗体侧经历了自己独立的进化,因此抗原表面缺乏与PPI相关的许多功能,包括缺乏非极性和芳香族残基的富集。这些界面具有较少的疏水性相互作用,通常由对位芳族热点组成,并被短链亲水性残基产生的极性接触所包围[5]。大多数交叉界面氢键是由侧链-侧链相互作用产生的,与obligate的PPI和酶抑制剂复合物相比,骨架-主链的氢键较少[6]。
三、什么是深度学习?
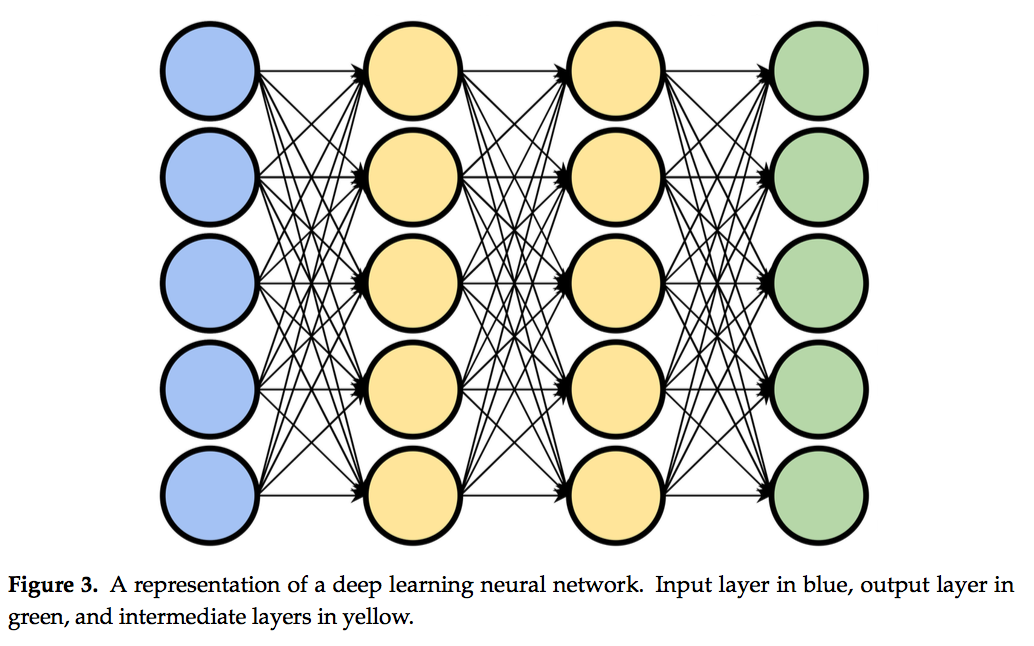
深度学习是机器学习的一个子集,它与算法特别相关,该算法特别能够从原始的低级数据表示中提取高级特征。这样的示例是一种算法,该算法从图像内的原始像素提取对象的曲率或深度。深度学习算法通常由具有一个或多个中间层的人工神经网络(ANN)组成。 ANN的中间层使网络“更深”,可以被认为负责将底层数据转换为更抽象的高层表示。网络的每一层都由节点或“神经元”的排列组成,每个节点或“神经元”都将一组加权值作为输入并将其转换为单个输出值(通常是通过对加权输入求和)。然后将结果值传递到后续层中的节点。第一层的输入值由从业者或模型设计师选择。在生化背景下,这些特征可以是手工制作的值(例如蛋白质的体积),也可以是较低级别的值(例如氨基酸序列)。在“深度”网络中,第一层的输出在产生最终结果的最终输出层之前经过一个或多个中间层。中间层允许网络通过依次提取更高级别的特征并将其传递到后续层来学习输入和输出之间的非线性关系(图3)。
为了使神经网络将输入转换为所需的输出,必须对其进行“训练”。训练神经网络的方法是修改节点之间的连接权重。在“完全连接”网络中,每个节点都连接到后续层中的每个节点。来自上一层节点的输出值通过加权连接传递到下一层节点。这些连接的权重通常最初是随机分配的,并且随着网络的训练,通过迭代地修改权重来进行校正,使得网络更倾向于从给定输入中产生所需的输出。模型的正确性由“成本函数”(cost function)确定,该函数提供了对模型输出中的错误量进行数值测量的方法。成本函数的选择在很大程度上取决于网络的任务,并用作最小化或最大化某些度量的代理,这些度量无法直接用于优化,因为它不可区分,例如分类精度。为了确定每个权重必须改变的方向以便更接近所需的输出,相对于网络的权重计算成本函数的偏导数[7]。成本函数的示例包括用于二进制分类任务的二进制交叉熵,或经常用于回归任务的均方误差。通过对整个数据集进行多次遍历重复此训练协议,可以对模型进行训练,以识别和权衡数据中可广泛预测最终结果的特征。像其他机器学习方法一样,使用训练,验证和测试数据集来评估模型性能。训练集是用于将模型增强到所需输出的数据的子集,而验证子集用于通过基于根据验证集计算的某些准则终止训练来防止网络过度拟合。一个普遍的标准是提早停止训练,一旦验证集的性能开始下降,训练就会终止。测试子集(通常称为保持集,hold-out set)用于通过对看不见的数据样本进行评估来分析训练后的模型的泛化能力。
在过去十年中,深度学习算法在竞争性任务(例如Go和Chess [8])中显示了超人的能力。 尽管这些方法受益于几乎无数的训练示例,但其他方法也已经看到了使用人工注释数据集的人工能力。 这样的应用包括图像分类和语音识别,其中手工制作的特征已被深度学习模型的内部层中提取的特征所取代。 虽然这些方法之间的应用领域与生物学有很大不同,但是这些应用程序中的数据表示与生物学数据之间存在相似性。 生物数据可能更复杂,并且这些方法学习丰富的高级功能的能力使其成为在更复杂的数据集中学习模式的有吸引力的方法。
3.1 建模(序列到结构) Modeling (Sequence to Structure)
蛋白质晶体结构在围绕蛋白质-蛋白质相互作用,蛋白质功能和药物开发的当前研究中发挥了作用。 尽管实验确定的蛋白质晶体结构的数量已显着增长,但与可用的序列数据量相比却相形见[[9]。 在过去的几十年中,随着测序技术的不断发展,已知蛋白质序列的数量呈指数增长。 由于这种差异,已经创建了几种三维结构建模方法,以弥合序列的可用性与已知结构的短缺之间的差距。
蛋白质结构建模的当前方法包括同源性建模和从头算建模。
- 在同源性建模中,将蛋白质的序列与具有已知结构的蛋白质的序列进行比较。 紧密相关的蛋白质或蛋白质结构域用作靶序列中相应区域的结构模板[8]。
- 从头建模用于没有相似序列的结构已知或不适用于同源性建模的情况。 在这种情况下,从头算术算法将尝试仅使用其序列来生成蛋白质的三维结构。 通常,这是通过对已知残基构象进行采样和/或搜索已知的蛋白质片段(局部蛋白质结构)以用作结构的一部分来完成的[10,11]。 这借助诸如基于知识和经验能量函数的工具来选择可行的结构。
3.2 接触预测/亲和力成熟/对接 Interaction Prediction/Affinity Maturation/Docking
抗体治疗剂针对靶蛋白而设计。因此,至关重要的是能够理解和推断抗体与靶标之间的结合行为,例如两种蛋白质是否具有能量上有利的相互作用(相互作用预测),哪些残基将形成相互作用界面,并且什么构象(对接),或某些氨基酸取代将如何改变结合能(亲和力成熟)。对接算法试图解决两个或多个交互结构之间的精确三维构象姿势。早在1982年就已经存在用于预测候选药物结合物的软件[12]。对接算法最初用于小分子配体,当前的标准包括GOLD,DOCK和AutoDock Vina,对接算法已扩展到蛋白质-蛋白质结构域,目前的标准包括ZDOCK,ClusPro,Haddock,RosettaDock等[13-16]。
这些方法的共同点是诸如蒙特卡洛(Monte Carlo)或快速傅立叶变换(Fast-Fourier transform)之类的采样技术,其目的是生成结构构象,该构象可以通过估算两个对接结构的能量偏好的函数进行评分[17,18]。可以使用基于能量偏好度对绑定结构进行分类的交互预测算法来筛选候选对象并缩小搜索空间。
另一组算法,称为B细胞反应的类似过程后的亲和力成熟,试图确定对结合配偶体的突变或修饰是否对结合亲和力或高能亲和力产生影响,或产生突变或序列,从而增加结合 伙伴亲和力。
3.3 目标识别(表位映射) Target Identification (Epitope Mapping)
目标识别包括在不了解蛋白质结合伴侣的情况下用于定位蛋白质结合位点的方法。 由于蛋白质对结合伴侣表现出特异性,因此这项任务相当困难。 抗体结合位点(表位)可分为两类:T细胞表位和B细胞表位。 B细胞表位可进一步分为线性和不连续[19]。 尽管T细胞表位的预测方法已经取得了更大的成功,但B细胞表位的预测仍然是一个困难且尚未解决的问题[20]。 尽管从理论上说这是一个无法解决的问题,但已经提出了几种方法,并取得了一定程度的成功[21]。
四、为什么要针对这些问题进行深度学习?
解决这些问题的传统方法往往依赖于
- 理论能量函数(基于物理学的) energy functions (physics-based)
- 或统计能量(基于知识)的能量。 statistical (knowledge-based) energies
但是,没有力场能够完全捕获与生物分子存在的复杂相互作用,基于物理学的模拟需要大量的计算时间。 深度学习提供了对当前能量函数无法捕获的属性执行高效的高级特征提取的功能。
近年来,由于几何深度学习和卷积神经网络,解决计算机视觉问题的解决方案的成功显着增加[22,23]。卷积神经网络以权重矩阵的形式利用跨输入域的滑动滤波器,该矩阵将输入转换为每个滤波器的滤波器匹配度。该滑动滤波器类似于正常网络层,该网络层仅从前一层中的神经元子集接收输入。使用这些技术在计算机视觉中看到的最新进展对于基于结构的深度学习方法特别重要,因为此类问题可以归结为计算机视觉问题。通过选择将蛋白质结构表示为例如图形,流形或3D体素网格,可以使其与这些卷积滤镜兼容。对于基于图的方法,原子或残基可以表示为节点,它们之间的相互作用采用边的形式。基于体素(Voxel-based)的方法将蛋白质离散成一个网格,并用组成成分(例如原子构成)标注网格点[24]。歧管方法(Manifold methods)可用于将蛋白质表面表示为整个结构域的相对曲率和能量特性。
此外,recurrent neural networks(通常用于语言处理任务中)可以通过将序列数据构建为具有唯一词汇的语言来加以利用[25]。 递归神经网络通常使用层的阵列状结构,其中层输入要么是直接输入值(例如单词中的字母),要么是隐藏输入,它们是数组中前一层的输出。 隐藏节点允许上下文沿数组传播,并在该上下文内评估后续输入值。
这些相似的深度学习方法在多个问题领域中的应用可以归因于它们学习基础表示的能力。 从抽象的意义上讲,不考虑基础架构的细微细节,仅当适用于一组数据时,未经处理且未经训练的深度学习模型才变得特定于领域。 但是,这种灵活性和对数据的依赖可以使模型对意外信号敏感(如对抗攻击所示)
五、深度学习方法
5.1 序列到结构 Sequence to Structure
5.1.1 抗体
改善抗体模型的努力主要集中在仅从其序列确定CDR的结构上。 诸如同源性建模之类的建模算法在确定非H3 CDR的结构方面已经取得了很大的成功,这些非H3 CDR大多属于规范结构簇,由关键残基的长度和氨基酸序列决定。 机器学习方法,例如梯度提升机(GBM)和位置特定评分矩阵(PSSM),已被用来学习如何将非H3 CDR分组和分类为结构簇[27,28]。 同一规范聚类中序列之间的强结构相似性使得对这些序列的建模相对简单。 使用高分辨率抗体蛋白质数据库(PDB)结构的精选集来完成这些模型的训练。
缺乏有效的建模方法以及H3 CDR的相对重要性,导致许多深度学习算法试图对H3 loop进行结构建模。这些方法之一是由Ruffolo等人开发的DeepH3 [29]。利用深度残基神经网络,DeepH3能够通过生成概率分布来预测残基间距离(d,使用Cβ原子和Cα为甘氨酸)和残基间定向角(θ,ω为二面角,而φ为平面角)在残基对之间。该模型的目的是查看由建模算法生成的H3 loop的假设结构,并对结构进行排序以识别H3的最可能构象。基准数据集来自PyIgClassify数据库(经过精心策划,包括去除冗余序列),仅包含人和小鼠的H3 [30]。为了进行训练,从结构抗体数据库(SAbDab)中提取了1462个结构,随机选择了5%的loo[并留待验证,以解决过度拟合的问题
DeepH3报告说,仅测量d和φ的两个变量(在这种情况下,预测角度与目标角度之间的相关性)之间的线性相关性的Pearson相关系数(r)分别为0.87和0.79,而圆形相关性系数(rc )(二面角ω和θ的圆形类似物Pearson相关系数)分别为0.52和0.88。将DeepH3与Rosetta Energy进行了比较,发现49个基准数据集结构平均提高了0.48Å。此外,他们能够显示DeepH3的分辨力得分(D,该模型区分好坏结构的能力)优于RosettaEnergy,分别为-21.10和-2.51。对于涉及两种抗体及其2800个诱饵结构的案例研究,DeepH3的一种抗体表现更好(D = −28.68,RosettaEnergy D = 3.39),而第二种抗体的表现稍差一些(DeepH3的D = 0.66和D = −1.59 RosettaEnergy)。
5.1.2 蛋白
AlphaFold
正如人们可能期望的那样,大多数用于建模生物分子的深度学习方法不仅专注于抗体,而且更广泛地专注于蛋白质-主要是在蛋白质折叠预测领域,该领域旨在从蛋白质的氨基酸序列生成结构。一种这样的方法是AlphaFold,其中通过使用PDB中的结构训练神经网络来预测残基Cβ原子之间的距离来产生蛋白质特异性电势[32]。初始预测后,可使用梯度下降算法将电势最小化,以实现最准确的预测。 AlphaFold使用从PDB中提取的结构的数据集,并使用CATH(类体系结构拓扑同源性超家族数据库)35%的序列相似性簇代表进行过滤,从而产生29,427个训练结构和1820个测试结构。以蛋白质结构预测的关键评估(CASP13)数据集为基准时,AlphaFold在所有组中表现最佳,可以为43个“自由建模域”(或没有同源结构的域)中的24个生成高精度结构。
AlphaFold方法的一个缺点是需要多序列比对,这可能会影响跨蛋白质的用途。
递归几何网络 Recurrent Geometric Network
AlQuraishi提出了只需要一个氨基酸序列并直接输出3D结构的深度学习方法[33]。 在这项工作中,循环神经网络被用来预测蛋白质骨架的三个扭转角。 AlQuraishi将他的方法,即递归几何网络(RGN)分为三个步骤。 在第一步中,RGN的计算单元将输入序列转换为代表每个残差的二面角或扭转角的三个数字,以及有关在相邻计算单元中编码的残差的信息。 该计算在整个序列中执行一次,然后执行一次,然后进行一次,从而允许模型创建整个蛋白质结构的隐式表示。
然后,在第二步中使用这三个计算出的扭转角在整个结构上一次构造一个残差。在最后阶段,通过将生成的结构与本地结构进行比较来检查生成的结构。所使用的得分是基于距离的均方根偏差(dRMSD),该偏差允许利用反向传播以优化模型。
在CASP11竞赛之前,所有可用的序列和结构数据都用于训练(保留一小部分用于验证和优化),而在实际竞赛中使用的结构则用于测试RGN。报告了自由建模(FM,新型蛋白质)和基于模板的建模(TBM,在PDB中具有已知同源物的结构)结构的结果,并将其与CASP11评估中所有服务器(自动化)组的结果进行了比较。在比较dRMSD值时,RGN的表现优于所有组,并且从FM类别的TM得分来看,RGN的总和最好。对于TBM,它没有击败前五名中的任何一个,但在dRMSD方面排名前25%。这些结果可以用以下优点和缺点来解释:使用dRMSD对模型进行了优化,在训练过程中从未看到过TM得分,并且不允许像其他小组一样使用基于模板的建模。
有趣的是,来自递归神经网络上游和下游计算的已解决扭转角在整个序列中的传播。 由于抗体构架区和非H3 CDR环的结构通常可以相对容易地建模,因此构成这些区域的残基的已解扭角可以轻松地在H3环的残基中传播。 上述方法的第一阶段。 实施这些修改所需的微小更改使其成为H3建模的有吸引力的框架。
变换约束的Rosetta(trRosetta) Transform-restrained Rosetta
预测残基间方向和距离的另一种方法是使用深度残基卷积神经网络。变换约束的Rosetta(trRosetta)使用输入序列和多序列比对,以输出预测的结构特征,将其提供给Rosetta构建协议以得出最终结构[34]。网络从PDB数据集中学习概率分布,并将此学习扩展到方向特征(残基之间的二面角)。经过高分辨率检查后,收集了30%的序列同一性,以及其他要求(例如序列长度和序列同源性),总共收集了15,051条蛋白链用于训练。使用来自CASP13的31个免费建模目标对网络进行了测试,并将其与建模评估中的顶级群体进行了比较。 TrRosetta的平均TM得分为0.625,超过了最高的服务器组(0.491)和最高的人员组(0.587)。使用来自连续自动模型评估(CAMEO)的131个“硬”和66个“非常硬”的目标进行了进一步的验证。对于“硬”(hard)设置,报告的TM得分(0.621)比Rosetta高8.9%,比前两组的HHpredB高24.7%。 “非常艰苦”的设定来自131个得分低于HHpredB的目标。这些结构的平均TM得分为0.534,比Rosetta高22%,比HHpredB高63.8%。 trRosetta研究小组指出,与挑战赛中的其他团队不同,trRosetta的测试并非盲目进行,他们计划在未来的蛋白质评估挑战赛中确认这些改进。最后,该小组研究了该网络在18种从头设计的蛋白质上的性能,发现他们的方法在预测设计的蛋白质结构方面比天然蛋白质的结构准确得多。
5.2。互动预测/亲和力成熟度 Interaction Prediction/Affinity Maturation
5.2.1 深度学习用于抗体前导优化
深度学习在相互作用预测领域的成功应用来自于基于序列的方法,该方法被Mason等人提出以优化现有的抗体候选物。 [35]。作者没有使用公共领域的数据集,而是通过向CDR的H3区引入突变并筛选与靶抗原结合的变异体来生成现有治疗性抗体的相对较少的变异体(5×10^4)。标记为结合或非结合的H3序列用作长期-短期循环神经网络和卷积神经网络的输入,这些网络经过训练可以预测序列的结合标记。然后,将训练有素的网络用于将7.2×10^7个候选序列的计算生成集合过滤为3.1×10^6个预测的binders。实验测试表明,在30种随机选择的预测结合序列中,有30种特异性地结合了靶抗原,其中30种之一表现出了三倍的亲和力增加。
通过基于结构的方法进行的比较分析突出了该方法的重要性。 作者证明结构建模软件生成的新结合序列建议的数量比实际预期的结合序列空间小几个数量级,并且建模结构的自由能估计不能用作结合活性的可靠分类器 。
尽管基于结构的方法有可能代表给定输入的更丰富的功能,但是由于已开发的实验方法(例如下一代测序),基于序列的方法受益于更多的数据可用性。
5.2.2。 恩斯·格拉德 Ens-Grad
另一个序列优化算法Ens-Grad使用神经网络和梯度上升的整体来优化H3种子序列[36]。 简而言之,Liu等报告称使用平移实验生成的实验噬菌体显示数据训练六个神经网络(五个卷积)的集合。 淘选实验(Panning experiments)通过使一组与噬菌体结合的H3序列经受结合竞争,从而使结合力得以富集,在该竞争中非结合剂被洗掉,结合剂被保留下来用于下一轮[37]。 使用均方误差损失进行富集回归,或使用二元交叉熵对H3 CDR分类进行了训练,对几种具有不同架构的不同模型进行了筛选,这些H3 CDR在淘选轮次中依次富集。
在拟合神经网络的整体之后,作者使用梯度上升来优化输入种子序列。 与梯度下降相反,梯度下降通常用于修改神经网络权重,以最小化诸如分类误差之类的损失函数,在这种情况下,使用梯度上升来修改输入序列,以使输出最大化。 作者建议,使用多个神经网络的集成,可以通过针对不同的网络输出进行优化来优化采取受控路径。
使用此优化方案,作者能够生成比种子序列和训练数据集中的序列具有更高富集性的序列。 这一重要结果表明,神经网络模型能够推断出超出输入训练数据的范围,这可能是通过学习确定富集结合的因素的高级表示来实现的。
此外,与更常见的生成模型(如变分自动编码器和遗传算法)相比,使用梯度上升方法的作者展示了优越的性能。 然而,尚不清楚这些方法之间的差异是否归因于优化风格,还是归因于网络架构的差异(例如,层数或层大小)。
与Mason等人开发的方法类似。 [35],这种方法完全避开了对结构数据的需求,而这种结构数据要难得多。 但是,这些方法不太可能在靶抗原之间很好地泛化。 在每种方法中,网络都适合于源自单个靶标抗原的数据点,因此将这种方法应用于不同的靶标将需要进行广泛的湿实验室测试,以生成训练数据并重新拟合模型。
5.2.3 DeepInterface
DeepInterface是一种基于结构的方法,旨在将处于对接构象状态的蛋白复合物分类为真或假结合物[38]。 网络的输入是一个体素网格,该体素网格由放置在接口周围的固定大小的框构成。 为了处理旋转歧义,作者将矢量在结构质心之间对齐到三个坐标轴之一。 网络本身由四个卷积层组成,然后进行批量归一化和整流线性单元。 为了将体素空间转换为一维向量并随后转换为绑定预测,将全局平均池应用于体素空间,然后是两个完全连接的层。
这里值得注意的是用于训练网络的负面数据的生成。 在这种情况下的负例是指不是真正粘合剂的任何结构。 使用否定示例是分类中至关重要的一步,因为网络必须暴露于某种形式的否定输入才能成功进行训练。 为了产生这些负面的例子,作者使用了基于快速傅里叶变换(FFT)的对接算法ZDOCK,从采样的构象集中选择了不正确的对接解[13]。
将蛋白质界面表示为体素网格(voxel grid)是一种直观但有问题的策略。
- 首先,网络的输入大小将体素空间限制为单个大小。作者通过将传递到网络中的接口的大小限制为足够小以适合边界网格空间的方式来克服这一问题。
- 其次,由于在所有界面上都没有公共轴,因此产生了旋转歧义问题。用于3D对象的类似体素方法通常可以利用隐式重力矢量来消除轴上的歧义。可以使用输入的随机旋转版本或通过将旋转池合并到网络体系结构中来完成其余轴之间的歧义处理。
但是,由于所需的可能旋转数呈指数增长,因此这些方法对于二维以上的应用是不切实际的。尽管有这些限制,DeepInterface在基准数据集上仍可实现75%的分类精度,这证明了该分类任务的可行性
由于先前提到的抗体-抗原界面和一般PPI之间的差异,因此尚不清楚此处介绍的模型是否能够避免假阳性分类。 当基础训练数据集不代表其实际用例时,此问题可能会导致分发失误。 但是,除了边界体素空间大小以外,DeepInterface中提供的模型体系结构和输入结构在某种程度上与所评估的接口类型无关。 但是,应该指出的是,模型对界面区域空间排列的依赖不应妨碍其对抗体-抗原界面的适用性,因为抗体-抗原界面在形状互补方面具有不可区别的差异。
5.2.4 MaSIF搜索
MaSIF方法来自不断发展的几何深度学习领域。从蛋白质表面的网格表示开始,通过选择网格上的一个点以及定义的测地线距离内(geodesic distance)的所有相邻表面点来创建贴片[39]。每个表面点都用几何和化学特征标注,这些特征描述了曲率,凹度,静电势,疏水性和氢键势。将补丁(patch)下采样到80个bin的网格中(5个径向×16角)。每个面元包含分配给相应面元的点的特征属性的统计平均值。通过极坐标和角坐标索引的80个bin作为输入传递到一组测地线卷积滤波器中,以生成蛋白质表面的一维描述符。旋转最大池用于克服角度模糊性。然后,通过完全连接的层完善一维描述符。其余的体系结构被认为是特定于应用程序的,它表示使用一维描述符作为特定应用程序模型的输入的能力。
为了训练用于相互作用预测的特定于应用程序的模型,使用了三重态损失函数(triplet loss function)的修改版本,该函数使锚点(结合蛋白补丁)的一维描述符与阳性(锚点的互补补丁)的一维描述符之间的欧几里德距离最小 ,并最大化锚点和底片(锚点的随机选择的非互补表面补丁)之间的距离。 如果斑块(patch)中心在蛋白-蛋白界面彼此之间的距离不远,则认为来自两个单独蛋白的两个表面斑为正对。
为了衡量模型的性能,作者使用几何和化学特征对相互作用对和非相互作用对进行了分类,并报告了接收器工作特性曲线(ROC AUC)0.99下的面积。 作者通过创建低,高和非常高的界面互补性子集,进一步评估了模型在数据的不同子集上的性能。 有趣的是,正如预期的那样,使用低形状互补子集上的几何和化学特征集,模型的分类性能下降到0.81 ROC AUC,而当仅使用几何和化学特征时,模型的分类性能下降到0.73和0.75 子集。
MaSIF搜索是针对抗体-抗原和蛋白质-蛋白质界面的混合训练的,两者之间没有区别。 如前所述,抗体-抗原相互作用显示出与其他蛋白质-蛋白质界面相似的形状互补性[6]。 该观察结果提供了与对DeepInterface进行调查时所期望的相似的证据,即在此方面能够捕获蛋白质-蛋白质界面之间几何匹配的模型应该很好地推断抗体-抗原界面。
严格地认为不是深度学习方法的其他机器学习方法进一步强化了这一点。 例如,开发了一种基于图的机器学习方法,称为突变截止扫描矩阵(mCSM,mutation Cutoff Scanning Matrix),它可以预测突变后亲和力的变化,并分别针对蛋白质-蛋白质和抗体-抗原突变进行评估[40]。 与蛋白质-蛋白质突变数据集mCSM-PPI拟合的模型比专门针对抗体-抗原相互作用的模型(Pearson系数为0.53)的效果(Pearson系数为0.35)明显更差。
5.2.5 TopNetTree
通过分析称为TopNetTree的深度学习方法,进一步增强了将抗体-抗原接口作为蛋白质-蛋白质接口的特殊情况的需要。 TopNetTree是一种最新的创新方法,使用来自持久同源性的技术作为将蛋白质结构表示为一组一维特征的手段。 具体而言,使用元素特定的持久同源性可以使拓扑特征特定于化学和组成特性,以及特定于突变位点内(或一定距离)的原子。 使用这些方法,可以提取一维条形码,这些条形码代表成对的原子相互作用,空穴的存在以及其他多原子结构(例如环)的存在。 除拓扑功能外,还包括其他几个功能,包括溶剂可及的表面积,部分电荷和静电溶剂化自由能。
通过串联从native和变异结构生成的特征来编码变异。 代表成对原子相互作用的第一级条形码用作卷积神经网络的输入,该卷积神经网络具有四个卷积层和一个退出(dropout)层。 对网络进行训练,以使最终输出与∆G之间的均方误差最小。 初始拟合后,将最终卷积层的输出logit馈入一组梯度增强树中,以对卷积特征的重要性进行排名。 最重要的特征与更高级别的拓扑特征相结合,作为最终一组梯度增强树的输入,以获得∆∆G的最终预测。
当在SKEMPI2数据库的子集上进行训练(不包括抗体-抗原复合物),并在抗体-抗原界面内的一组787突变进行测试时,TopNetTree的Rp为0.53,均方根误差(RMSE)为1.45 kcal mol-1 [42]。 当对上述仅包含蛋白质-蛋白质界面的训练集进行十倍交叉验证时,作者报告的Rp为0.82,RMSE为1.11 kcal mol-1。 与突变后亲和力变化的其他预测因子进行比较时,TopNetTree展示了常规蛋白质-蛋白质界面以及抗体-抗原界面的最新技术成果。 蛋白质-蛋白质突变与抗体-抗原突变之间的∆∆G可预测性之间的性能差异突出表明,有必要将抗体-抗原界面视为单独的特殊条件。
5.3。目标识别
5.3.1 抗体特异性B细胞表位预测
与交互预测方法类似,可以根据所使用的输入将目标识别方法分为两个主要类别:结构式或顺序式(structural or sequential)。我们在这里首先回顾的是一种结构化方法,该方法证明了在没有关于相互作用抗体对位的信息的情况下预测抗原表面上相互作用域的挑战越来越大[43]。在Jespersen等人的这项工作中,为了制定可以输入到完全连接的神经网络层中的一维输入向量,作者首先将贴片(patch)定义为残基,并将其所有表面暴露的邻居定义为残基。接近6 A为了表示补丁的几何特性,作者使用了Cα原子和Zernike矩的前三个主要成分。 Zernike矩在这项工作中特别值得注意,因为它们的功能类似于卷积神经网络的过滤器,它通过将基础补丁解卷积为表示补丁中发现的特定形状和图案的程度的标量值。除了这些几何特征外,还包括诸如溶剂暴露和氨基酸组成统计信息等组成特征
对于训练数据,作者通过随机选择一个非表位残基并通过蒙特卡洛方法生成一个补丁来构建负补丁,该蒙特卡罗方法可反复向该补丁中添加相邻残基,并从中去除相邻残基。 斑块(patch)的目标值介于0和1之间,由与已知真实表位重叠的残基量决定。 同样,通过将表位与来自不同抗体-抗原簇的表位相匹配,可以产生负的表位-表位补丁对。
使用了三种模型:完整模型,最小模型和抗原模型-每个模型都具有两个隐藏层和一个S型激活函数,并且仅输入层的大小不同。 完整模型和最小模型都使用来自表位和对位的补丁特征,并经过训练以对配对进行评分,而抗原模型仅使用表位特征并且经训练仅对表位进行评分。 与完整模型相比,最小模型不包含Zernike矩的复杂结构特征。
为了比较这三种模型,作者使用上述方案针对八个不同抗体/抗原簇中的每个真实表位或表位/互补位对构建了300个阴性样品的测试集。使用模型评分对301个聚类进行排名,并确定Frank评分作为排名高于阳性的阴性样本的百分比。对于完整模型,最小模型和抗原模型,报告的分数分别为7.4%,10.9%和15%。这些结果清楚地证明了在有和没有候选抗体信息的情况下预测表位之间可行性的差异。尽管排除Zernike矩不能直接归因于完整集和最小集之间的性能下降(由于差异集中包含其他特征),但结果确实提供了证据,表明将表面斑解卷积为简单图案的组成就像在卷积神经网络中经常看到的那样,在处理结构数据时可能是一个强大的工具。
5.3.2 MaSIF-Site
如前所述,一种这样的方法实际上采用了上述的表面反卷积方法。 也就是说,MaSIF方法旨在使用测地卷积层从表面斑块生成一维指纹[39]。 上面的“交互预测”部分5.2.4中概述了MaSIF方法。 如前所述,这些指纹可以被馈送到特定应用层。 MaSIF站点就是这样一种应用程序。 与MaSIF搜索相反,作者报告了通过堆叠两个或三个测地卷积滤波器的层来进行不同网络深度的实验。
而且,与MaSIF搜索相反,作者没有提供几何和化学子集下的性能实验结果。 但是,据报道,用于预测相互作用和非相互作用斑块的模型分类性能的ROC AUC为每种蛋白质0.87 ROC AUC。 作者还通过评估模型对疏水性大的蛋白与疏水性小的蛋白的分类性能,给出了更细致的结果。 对于大疏水性,报告的性能为0.87,对于较小疏水性斑块,为0.81。 这在表位的情况下意义重大,因为抗体-抗原界面的疏水相互作用往往比一般的蛋白质-蛋白质界面少。 但是,该模型在将野生型非抗原蛋白补丁与具有已知抗体结合剂的突变版本区分开来时显示出令人满意的结果,表明其可用于鉴定表位。
5.3.3 线性B细胞表位
虽然据估计大约90%的B细胞表位是构象的,但人们对预测线性B细胞表位的关注度很高[20]。 Saha等人建立了用于预测线性B细胞表位的第一个神经网络模型。 [44]。 Saha等人使用相对标准的递归神经网络架构,该架构以氨基酸序列作为输入。 报道说,尽管训练了700个序列,但是从随机选择的线性表位残基(大概是非表位残基)分类中,预测准确性为65.93%。
线性B细胞预测的另一种直接架构使用固定大小为20的长度序列作为具有两个隐藏层的全连接架构的输入,并使用最终的softmax输出最终将输入序列转换为0到1之间的概率得分。 与其他应用程序一样,由于使用了不同的数据集,因此很难直接将此模型与前面提到的模型进行比较。 但是,报告的分类准确性为68.33%确实建议改进。
将这些方法彼此之间以及与其他非深度学习表位预测因子进行了更好的比较,同时引入了另一种深度学习模型,该模型被Sher等人开发,称为深岭回归表位预测因子(DRREP,deep ridge regressed epitope predictor )。 (表1)[45]。 简而言之,模型的初始层使用了一组随机的k-mer,该k-mer跨输入序列滑动,并用于计算每个k-mer和整个输入序列的子序列的匹配得分。 包含第二个合并层使此过程类似于卷积步骤,在卷积步骤中,将滤波器预设为随机k-mers。 与在神经网络训练中最常用的方法相反,第三层(完全连接的层)的权重是使用岭回归来分析计算的。 最后,输出层用于提供序列中每个残基的残基水平预测。
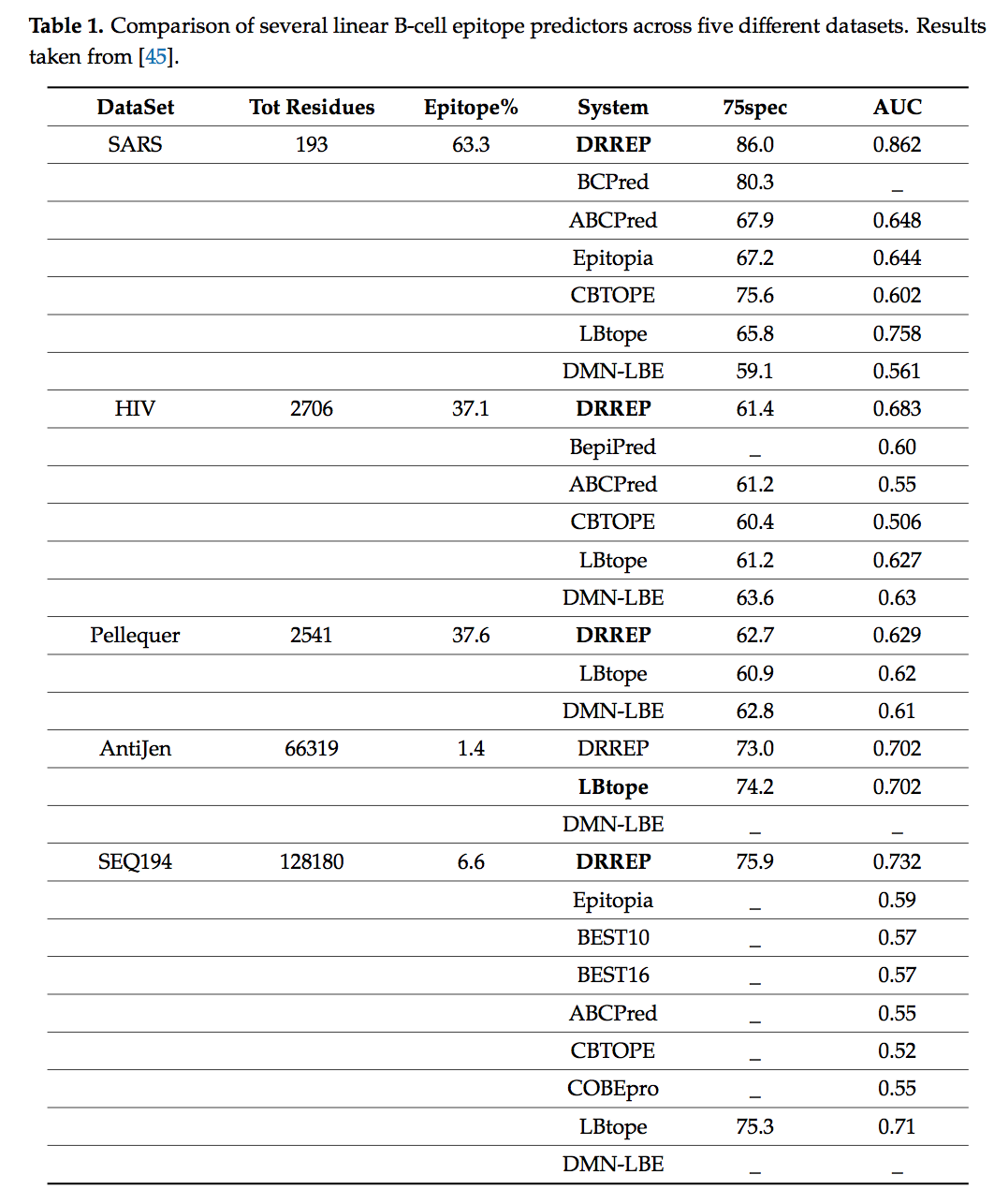
使用五个不同的数据集对上述方法进行基准测试。 在每种情况下,除了AntiJen数据集上基于支持向量矩阵(SVM)的LBTope之外,DRREP都展示了所有深度学习方法中的最佳性能,以及所有其他预测变量中的最佳性能。
六、数据集/基准 Datasets/Benchmarks
6.1 AB-Bind
AB-bind数据集是针对32种不同的抗体-抗原结构的1101个突变的集合,包括实验确定的与每个突变体相关的结合自由能的变化,以及测试每个突变体的实验条件[46]。 提供每种天然蛋白质复合物的结构时,该数据库的用户可以对突变引起的天然结构构象的任何变化建模。
6.2 AntigenDB
AntigenDB是包含结构,序列和结合数据的经过验证的抗原数据库[47]。 与其他抗原数据库相比,AntigenDB包含有关经过验证的抗原的数据,即使没有基础表位也是如此。
6.3 AntiJen
另一个抗原数据资源是AntiJen数据库,它是B细胞和T细胞抗原的精选数据集,具有实验注释,指向已发表的实验文章的链接和PDB条目[48]。
6.4 CAMEO
连续自动模型评估(CAMEO)平台通过每周进行全自动盲预测评估来补充CASP [49]。 挑战使用预发布的结构序列完成,这些序列将在其下一个发布版本中发布到PDB。 每五周平均总共有100个目标,开发人员能够更频繁地进行基准测试和验证其方法,这使其他小组可以访问更多基准测试结果数据。
6.5 CAPRI
预测相互作用的关键评估(CAPRI)是一项年度竞赛,邀请参加者提交未公开晶体结构的蛋白质相互作用的结构模型预测[50]。 然后根据球队的准确性对球队进行排名。 在结构建模预测中,许多论文经常通过与最近的CAPRI竞赛中表现最佳的竞争对手进行比较来测试其算法。
6.6 CASP
结构预测技术的关键评估(CASP)是两年一次的竞赛,邀请参与者为未发表的晶体结构提交结构建模预测(仅根据序列)[51]。 然后根据球队的准确性对球队进行排名。 许多从事结构模型预测(结构顺序)的论文都通过与最近的CASP竞赛中顶级竞争对手的表现进行比较来测试其算法。
6.7 DOCKGROUND
DOCKGROUND是蛋白质-蛋白质复合物的基准数据集,以及对接算法GRAMM-X产生的诱饵[52]。 数据集由61个真实复合物以及100个生成的诱饵(负数)组成。
6.8 免疫表位数据库(IEDB,The Immune Epitope Database)3.0
IEDB包含有关973,072个线性和不连续表位的序列,实验数据以及在某些情况下的结构数据的信息[53]。 IEDB的当前过滤搜索提供了来自826个抗原的4416个不连续表位。 其中,1318个表位具有已解析的3D结构。 线性表位的数目多得多,总计968,656。 但是,其中只有961个具有可解析的3D结构。
6.9 整合的蛋白质-蛋白质相互作用基准
包含蛋白质复合物,可以解决其结合和未结合的结构,其X射线结构的分辨率小于3.5 A ,并且是非冗余的[54]。 数据细分如下:总共230种复合物(40种抗体/抗原),其中179种包括亲和力测量(33种抗体/抗原)。 基于亲和力,数据集进一步分为三类,强调低亲和力的复合物代表了更具挑战性的数据样本。
6.10 PDB
蛋白质数据库可能是最大的结构解析生物分子数据库[55]。 它是由全球研究人员提供的,并由国家科学基金会(NSF),美国国立卫生研究院(NIH)和能源部(DOE)资助。 该数据库是许多精选数据集的来源。
6.11 PDBbind
每年更新一次,PDBbind数据库目前包含从PDB提取的2594种蛋白质-蛋白质复合物的已解析结构,以及每种复合物的结合亲和力数据[56]。
6.12 PIFACE
PIFACE数据集是一组独特的蛋白质-蛋白质复合物[57]。 使用结构相似性和基于图论的聚类方法对数据集中的聚类进行聚类,以识别22,604个唯一interfaces。
6.13。 PPI4DOCK
PPI4DOCK是另一种蛋白质-蛋白质复合物数据集,具有通过对接算法ZDOCK生成的诱饵[58]。 此数据集中的结构来自处于未绑定状态的同源性建模结构,以及每个复合物的实验确定结构。 使用CAPRI标准将数据集中的诱饵标记为“不正确”,“可接受”,“中等”或“ “高质量。
6.14 SAbDab
SAbDab是结构抗体数据库,每周自动更新一次,并包含诸如亲和力数据之类的注释,以及诸如CDR分类之类的其他抗体特异性注释[31]。
6.15 SKEMPI
SKEMPI数据库是蛋白质突变的策展,这些蛋白质的突变已在结构上得到解决,并且包括结合自由能的变化。 在撰写本文时,数据库包含7085个条目[42]。
七、讨论
如整篇文章所述,应将抗体-抗原相互作用作为蛋白质-蛋白质相互作用的特殊情况来对待。 一些研究通过提供它们在一般蛋白质和抗体特异性数据集之间的模型比较,直接加强了这一点。 其他方法则间接地强化了这一点,例如使用MaSIF方法,该方法表明具有较小疏水区域的蛋白质界面(抗体-抗原界面的特征)更难以分类[40,56]。 但是,使用抗体确实具有一些优势。 六个CDR环中的五个具有规范结构,提供了从环序列推断结构的可靠方法,并将建模工作范围缩小到了H3区域。 此外,结构特性和结构使得定义抗体的共同轴成为可能,从而潜在地解决了结构表示中出现的旋转歧义(rotation ambiguity)问题[59]。
尽管对于每个问题域都有几个常用的数据集,但并非所有方法都利用一个公共集。 此外,几种方法使用给定数据集的精炼形式或不同协议对训练,验证和测试数据进行聚类和拆分。 这些差异使得很难对方法进行精确比较。 我们希望本文中提到的数据库的管理能够使该领域更接近其他领域的地位,例如图像识别。 随着数据和方法数量的不断增长,维护基准的计划(例如CASP和CAPRI)将变得越来越有价值
深度学习需要大量数据。它在大量数据可用的域中的卓越性能证明了这一点。尽管目前确实有大量的蛋白质序列数据可用,但结构数据仍然滞后-由于既要通过实验解决蛋白质结构的费用,又要从序列中提取结构很困难。由于抗体-抗原结构是蛋白质复合物的一个子集,因此抗体结构数据的数量甚至更加有限。尽管未在上述任何方法中应用,但克服有限的抗体-抗原数据的一种策略是利用转移学习。转移学习是将学习过程中获得的知识从一个问题领域转移到另一个问题领域的过程[60]。例如,由于蛋白质-蛋白质相互作用受与抗体-抗原相互作用相似的生化原理支配,但表现出不同的潜在相互作用分布,因此网络最初可以适合蛋白质-蛋白质数据集,随后可以通过专门的抗体-抗原数据集来完善。这种方法允许网络捕获控制这两种现象的高级原理,同时为抗体特异性应用留下完善模型的空间。该技术的先前示例是使用蛋白质折叠网络,该蛋白质折叠网络在应用于膜蛋白问题之前,已在普通蛋白上进行了训练
由于它们的进化,许多蛋白质共享结构和序列相似性。工具和算法(例如用于序列相似性的BLAST或HMMER和用于结构相似性的FATCAT)提供了一种基于相似性指标对蛋白质进行评分(并因此进行聚类)的方法[62-64]。一些数据集提供聚类集,而其他数据集则将其留给用户以生成聚类。聚类并使用每个聚类中的代表性样本可以确保高度相似的潜在聚类成员不会分布在训练,验证和测试集中。这是任何基于学习的方法中的重要步骤,可减轻由于过度拟合导致的数据泄漏或夸大结果。蛋白质结构数据可以通过几种方法获得,但最常用和最准确的方法是X射线晶体学。尽管它倾向于提供比其他方法更好的分辨率,但是它并不完美。晶体学实验结果的准确性可能取决于所用设备和溶液的类型以及蛋白质本身的固有结构和稳定性。具有低分辨率的结构在代表真实蛋白质结构方面通常不那么值得信赖,并且许多具有“未解决”的区域,在这些区域中没有可靠的结构数据,尤其是在高度灵活的区域内,例如上述的H3环
此外,亲和力测量对实验设置敏感,实验设置可能因实验而异。 这可能是有问题的,因为此信息通常不包含在用于预测这些值的模型输入中。
必须克服的一个障碍是针对分类问题生成有意义的否定数据(negative data )。例如,关于靶标选择:尽管蛋白质与蛋白质之间的界面非常保守-因此,非相互作用残基可以被如此可靠地标记-抗体理论上实际上具有结合靶标蛋白质上任何区域的能力。因此,否定标签是一个棘手的问题。换句话说,缺乏显示抗体与特定区域结合的数据不足以证明抗体不可能与该区域结合。这意味着标记的抗原残基尚未确定与任何抗体相互作用,因为它们肯定是“非相互作用的”,可能会导致分类错误。此外,即使旨在将界面或复合物分类为结合剂或非结合剂的方法也必须小心如何产生负结构。例如,DeepInterface对接诱饵的使用意味着对分布失调预测的任何依赖都取决于对接诱饵的质量。也就是说,较差的对接构象可以很容易地与真正具有约束力的构象分开,因此,需要对实际上无限的负空间进行大量采样。但是,诸如MaSIF中使用的那些技术(其中两个复合物彼此之间的空间方向可以完全忽略)不会遭受相同的风险。
随着深度学习的领域继续扩展到其他领域,新颖的体系结构和表示输入数据的方法很可能随之而来。这些领域将带来新的任务,这些任务可以重新格式化并与生物学应用相关。例如,MaSIF方法中使用的三重态损失(triplet loss)函数以前曾用于人脸识别[66]。至于数据表示,体素网格(voxel grids )以前用于对象分割和识别[67]。同样,递归神经网络被用来分析书面文本[68]。确定这些新颖方法是否将适用于生物制品领域的一种策略是评估以与开发体系结构兼容的格式表示生物数据的可行性。在这项工作中,我们专注于序列和结构数据。但是,生物学数据不限于这些表示。例如,蛋白质之间的相互作用或蛋白质属性之间的相似性可以表示为图网络。
总而言之,为了使这些生物学数据适应开发架构的使用,必须首先建立兼容的表示形式,并将其映射为适当的格式,然后严格评估表示形式中的哪些模式可以被深度学习框架捕获。
7.1 未来发展方向
7.1.1 结构表示 Structural Representations
特别是对于基于结构的方法,数据表示方案的选择在应用深度学习方法中起着重要作用。 不断增长的几何深度学习领域提供了许多示例,其中以局部为基础的坐标系统中的结构数据表示既降低了模型的复杂性,又为诸如旋转的变换提供了不变性。 此类方法还规避了基于体素(voxel-based)的技术中容易出现的问题,例如数据稀疏性(大多数体素包含空白空间)和对输入的严格约束(例如体素网格的边界大小)。
7.1.2 标准数据集和基准 Standard Datasets and Benchmarks
在任何深度学习领域中开发和改进新方法的关键是要有一个通用的基准数据集。 例如,小分子设计中的深度学习应用受益于常用的基准数据集,例如ZINC和ChEMBL,基于图像的深度学习方法经常在MNIST或ImageNet上进行评估[69,70]。 这样的数据集的特征在于大量的数据样本,因此可以对训练,测试和验证集进行拆分。 随着该领域的不断发展和附加数据量的增长,希望将出现大型标准数据集,这些数据集可用于直接比较模型架构和技术并提供更清晰的前进方向。
7.1.3。 生成方法 Generative Methods
来自小分子发现和设计领域的一种尚未深入到生物学的深度学习方法是将复杂的生成框架用于结构生成。 已经提出了几种方法,它们可靠地产生新的稳定的化学结构,甚至允许实施特定的化学性质,但是这些方法不适用于蛋白质。 与小分子相比,这种差异可归因于蛋白质结构的复杂变化的大小和数量更大。 适用于生物制剂的生成模型的开发可能会导致通往能够直接基于给定epitope设计paratope结构和组成的网络。
7.1.4 分子动力学的深度学习
在这篇评论中没有涉及深度学习与分子动力学的交叉发展。 简而言之,分子动力学模拟旨在重现现实中表现出的原子运动。 分子动力学的应用广泛,该领域内的革命可以加速许多蛋白质工程任务的发现和发展。 有关涵盖该领域深度学习进展的详细信息,请读者阅读有关机器学习和分子动力学[71]主题的评论文章。
八、结论
本文讨论了使用过去几十年来创建的大量生物学数据来增进我们对蛋白质生物化学理解的几种方法。 深度学习已成为整个生物化学科学研究不可或缺的一部分,并且日益成为药物发现工作的组成部分。 作为研究对象的生物药物(尤其是抗体)的增长明确表明,必须采用深度学习方法来改善该领域药物发现工作的成果。 抗体药物具有以高亲和力和特异性结合其临床靶标的能力,并且显然需要专门适应于理解和增强这些蛋白质的机器学习算法。
但是,在可用蛋白质数据的数量和类型方面仍然存在重大挑战。尽管确实存在大量可用的蛋白质结构,但由于现代测序技术所提供的序列数据的激增,数据量却相形见绌。新的蛋白质结构求解方法的开发可能会改善这种差异,但就目前而言,相对较小的结构数据量限制了某些深度学习方法的有效性。此外,可能很难找到负面数据,尤其是对于绑定预测等问题,在这种情况下,传统的结构确定方法只能产生正面数据。通过计算生成此类负面数据会带来一系列挑战。很难知道生成的负片是否确实是真正的负片,而生成看起来足够逼真的负片也同样困难。即使在很好理解的肯定数据的范围内,这些模型中的每一个使用的各种训练数据集的不同性质也使它们难以比较,并且难以确定哪种方法对于给定的应用是最佳的。
随着新数据和方法的出现,以应对当前深度学习及其在蛋白质科学中的应用的局限性,在我们对这一充满挑战的空间的理解中,我们将继续看到越来越多的进步。 将这些改进成果转化为对抗体药物开发的新认识,对于创建和改进新药物至关重要。 深度学习与抗体科学的结合具有无穷的潜力,可以帮助创造新的,先进的药物,从而突破当前技术的界限。
参考资料
- A Review of Deep Learning Methods for Antibodies.Antibodies 2020, 9, 12; doi:10.3390/antib9020012
