【4.5.2】I-TASSER服务器--蛋白质结构和功能预测的新发展
I-TASSER服务器( http://zhanglab.ccmb.med。umich.edu/I-TASSER )是用于自动化蛋白质结构预测和基于结构的功能注释的在线资源。在I-TASSER中,首先使用多线程对齐方法从PDB识别结构模板。然后,通过迭代片段装配模拟来构建全长结构模型。通过将预测的结构模型与功能数据库中的已知蛋白质进行匹配,最终得出功能信息。尽管该服务器已广泛用于各种生物和生物医学研究,但用户社区已报告了许多评论和建议。在本文章中,我们总结了I-TASSER服务器上的最新发展,旨在满足用户群体的需求并提高建模预测的准确性。重点介绍了用于原子级结构细化,局部结构质量估计和生物功能注释的新方法。我们希望这些新的发展将提高I-TASSER服务器的质量,并进一步促进社区对其进行高分辨率结构和功能预测的使用。
一、前言
随着蛋白质结构预测的进展,分子和细胞学研究人员已成为在进行实验研究之前寻求其蛋白质自动服务器预测的常规方法。 整个社区的blind CASP实验表明,许多自动化服务器(例如I-TASSER(1,2),Rosetta(3)和HHpred(4))现在可以生成结构模型,其准确性可与最佳的人类专家指导建模进行比较。。 从计算结构模型开始,已经提出了许多方法来注释蛋白质分子的生物学功能,例如包括COFACTROR(8,9),COACH(10),Con-Cavity(11)。 ,FINDSITE(12),Firestar(13)和3DLigand-Site(14)等。 最近已经开始使用基于活动平台的平台来评估配体结合位点预测(15),这证明了自动化蛋白质功能注释的准确性和实用性(16)。
I-TASSER服务器代表了用于自动化蛋白质结构预测和基于结构的功能注释的最广泛使用的在线系统之一(1,17)。 从氨基酸序列开始,I-TASSER通过重新组装从threading模板上切除的片段来构建3D结构模型,在该模板中,通过将结构模型与功能数据库中的已知蛋白质进行匹配,可以得出目标蛋白质的生物学见解(18)。 自2008年首次变身(incarnation)以来(17),I-TASSER服务器系统已经为117个国家的5万多名用户提交了超过20万种蛋白质的全长结构模型和功能预测。
尽管得到了广泛的使用,但我们还是从用户社区收到了许多有关改进系统的意见和建议。在本文中,我们报告了I-TASSER服务器的最新发展,这些发展显着提高了I-TASSER模型的质量和服务器系统的功能。主要的新进展包括:(i)估计结构模型残基水平的局部质量的新方法,这对于指导生物学家用户进行功能研究至关重要; (ii)B因子预测算法; (iii)改进原子级结构的方法,以改善I-TASSER模型的氢键网络和物理真实性; (iv)基于共识的配体结合位点预测,结合了COACH的结构和序列特征比较(10); (v)新功能库BioLiP(18)的集成,以增加蛋白质功能注释的覆盖范围; (vi)开发新的留言板系统,以促进与用户社区的讨论和交流。
二、材料和方法
2.1 I-TASSER管道概述 Overview of the I-TASSER pipeline
I-TASSER服务器(http://zhanglab.ccmb.med.umich.edu/I-TASSER/ )建立在I-TASSER上,I-TASSER是一种基于分层模板(hierarchical template-based )的蛋白质结构和功能预测方法,包括三个常规步骤(图1)。
- 对于给定的查询序列,I-TASSER首先使用LOMETS(20)识别PDB库(19)中的结构模板或超二级结构模体,LOMETS(20)是由多个线程算法组成的元线程程序。
- 然后,通过重新组装从模板中切除的连续排列的片段结构,构建全长模型的拓扑结构,其中,未复制区域的结构是通过基于副本交换蒙特卡洛模拟的从头开始折叠从头开始构建的( 21)。 SPICKER(22)对结构轨迹进行聚类,以识别低自由能状态。从SPICKER集群开始,进行了第二轮结构重组以完善结构模型。通过使用FG-MD(23)和ModRe ner(24)进行全原子模拟,可以进一步降低低自由能构象。
- 最后,通过结构和序列特征比较,通过将结构模型与BioLiP功能库中的蛋白质匹配,可以获得查询蛋白质的功能见解(8,10,18)。
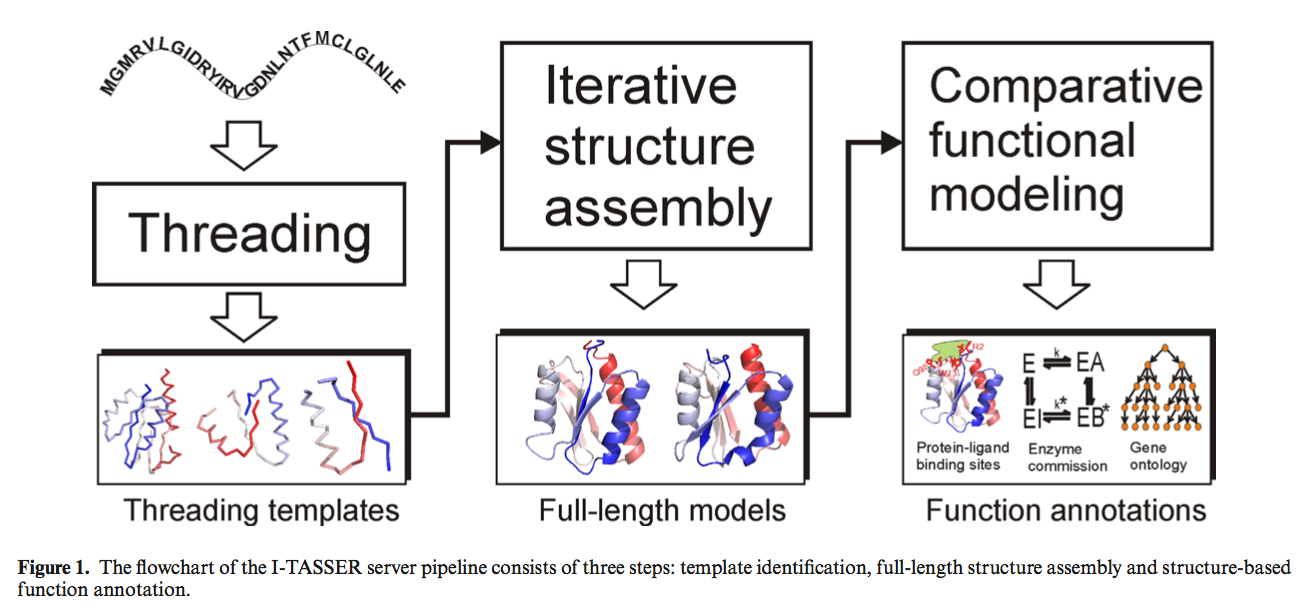
I-TASSER管道与在CASP实验中Zhang-Server使用的方法相同。 然而,自从CASP9以来,一种新的从头开始结构预测方法QUARK(25)已被引入到Zhang-Server管道中,以识别和排序硬自由建模(FM,hard free modeling)目标的模板(26,27)。 QUARK程序尚未集成到I-TASSER服务器中,该服务器可作为独立的在线服务器从头开始折叠目标,位于 http://zhanglab.ccmb.med.umich.edu/ QUARK/ 。
2.2 I-TASSER服务器的最新发展
估算I-TASSER模型残基水平局部质量的新方法。
I-TASSER提供了相关分数(C分数)以估计模型的整体准确性。但是,在以前的I-TASSER结构预测流程中,对基于结构的功能研究(例如活性位点识别,配体-蛋白质对接和药物筛选)至关重要的残基水平的局部准确度。为了解决这个问题,我们开发了一种新算法ResQ(Yang等,已提交),用于估计I-TASSER模型的残基特定局部质量。 ResQ考虑了多种信息来源,包括:(i)蒙特卡洛装配模拟的结构变化,(ii)通过穿线和结构对准检测到的模板的距离偏差,(iii)threading对准覆盖率和(iv )模型与基于序列的结构特征预测之间的一致性。
ResQ对残基的质量进行了评估,评估了一组非冗余的635种蛋白质,其结构模型由I-TASSER生成,其中I的估计和观察到的距离误差之间的平均差异-TASSER模型的C评分> -1.5的506种蛋白质为1.4Å。 ResQ方法还通过在CASP9和CASP10实验中由各种预测变量生成的结构模型进行了测试。结果表明,ResQ与局部结构质量评估的模型质量评估程序(MQAP)相当或优于大多数模型质量评估程序(MQAP)(请参见补充表S1-S4)。 ResQ程序可用于评估由I-TASSER和其他结构预测方法生成的结构模型的准确性。可以通过位于 http://zhanglab.ccmb.med.umich.edu/ResQ/ 的在线服务器进行访问。 或下载作为独立软件包 http://zhanglab.ccmb.med.umich.edu/I-TASSER/download 。
B因子预测 B-factor prediction
结构预测流程中另一个高度相关但经常被忽略的局部特征是蛋白质中残基的固有热迁移率。在绝对零温度下,蛋白质中的原子被推定为处于最低能量的平衡位置。但是随着温度的升高,环境热能使原子在平衡位置附近振荡,其程度通常取决于3D结构上的相对位置以及与配体和溶剂原子的相互作用而变化。原子运动可以在X射线晶体学中作为B因子(或温度因子)进行实验测量,作为对结构因子方程的修正因子而引入,因为X射线对振荡原子的散射效应降低了与静止原子相比(28)。由于蛋白质晶体中热运动因子的分布会受到系统误差(例如实验分辨率,晶体接触和修饰程序)的影响,因此原始的B因子值通常在不同的实验结构之间无法比较。因此,为了减少影响,我们使用基于Z分数的变换来计算归一化的B因子。在ResQ中使用结合了序列分析和预测结构特征的基于模板的分配和基于机器学习的预测相结合来预测归一化的B因子(Yang等,已提交)。对上面提到的635种蛋白质的测试表明,与X射线晶体学数据相比,ResQ估计的B因子在曲线下的平均面积(AUC)为0.79(请参阅补充表S5)。
原子级结构修饰 Atomic level structure re nement
在以前的I-TASSER建模版本中,最常见的投诉之一是结构模型的局部结构质量。例如,即使总体拓扑通常是正确的,某些模型仍具有不切实际的二级结构特征或空间冲突。这通常发生在非同源蛋白靶标上,在这种情况下,结构装配模拟由于线程排列不一致而趋向于产生不同的诱饵(decoys)。因此,来自不同诱饵(decoys)结构的组合结构模型会导致局部结构图案失真。
为了提高局部结构质量,我们开发了两种算法FG-MD(23)和ModRe ner(24)。在FG-MD(23)中,我们通过模拟退火分子动力学模拟来放松I-TASSER模型,其中将PDB库中通过TM-align检测到的结构片段的距离图用作空间约束。在ModRener(24)中,结构模型分为两个步骤:
- 首先通过氢键优化从C迹线构建骨架结构,
- 然后使用快速蒙特卡洛模拟重新包装侧链旋转异构体。基于物理的原子力场。
两种方法已集成到I-TASSER管道中,以定义不同目标的模型。通常,FG-MD用于带有同源模板的易标靶; ModRener被用于没有同源模板的硬目标,以生成完整的原子模型,随后是FG-MD修正。基准测试和CASP测试的数据表明,I-TASSER模型的HB得分(测量氢键网络的质量)和Molprobity得分(测量空间碰撞和Ramachandran规律性)可以显着提高re nement模拟(27,29)。
配体结合位点预测的共识方法 Consensus approach to ligand-binding site prediction.
I-TASSER中的配体结合位点以前是由COFACTOR预测的,它可以通过整体和局部结构比较检测到的同源模板推导结合位点(8)。但是,我们发现,仅凭一种基于结构的比较的方法就可能因开发而导致缺少一些有用的功能模板。结果,我们开发了两种新方法,一种基于结合特异性子结构比较(TM-SITE),另一种基于序列特征比对(S-SITE),用于互补结合位点预测。将TM-SITE和S-SITE与COFACTOR,ConCavity(11)和FINDSITE(12)结合使用时,开发了一种称为COACH的共识方法,该方法显着改善了我们大规模基准测试中的任何一种方法(10 )。我们认为,结合其他互补配体结合位点预测方法,例如Firestar(13)和3DLigandSite(14),将进一步提高COACH方法的性能。
集成了新功能库BioLiP。
I-TASSER基于结构模型与功能库中模板的匹配,对查询蛋白的生物学功能进行注释,包括配体结合位点,酶促(EC)数和基因本体(GO)术语。 PDB库(19)是用于研究蛋白质结构和功能的主要信息库。但是PDB中的许多蛋白质都包含冗余条目,残基排序不正确和功能信息不明确。特别是,许多蛋白质是使用人工分子作为添加剂来解析的,以利于结构测定。这些问题使PDB不能成为进行基于结构的精确功能分析的可靠资源。已努力解决这些问题,以建立与生物学相关的配体-蛋白质结合数据库,例如FireDB(30)。
为了评估每个PDB条目的生物学相关性,我们提出了一个分等级的程序,该程序由基于计算机的过滤和手动文献验证的四个步骤组成。因此,该程序用于从已知蛋白质结构/功能的数据库和PubMed的文献中构建一个综合的功能数据库BioLiP(18)。除了配体结合位点和相关的结合能力数据外,BioLiP中的每个条目还包含EC号和GO术语,它们均每周更新一次,可在 http://zhanglab.ccmb.med.umich.edu/BioLiP 上免费获得。
当前版本的BioLiP(2015年1月16日)包含304050个条目,该条目由65506个PDB蛋白构建而成,其中37685个条目用于DNA / RNA-蛋白相互作用,13967个条目用于肽-蛋白相互作用,85652个条目用于金属离子-蛋白质相互作用,而常规小分子-蛋白质相互作用为166 746。它涉及76 881条蛋白质链,分别具有518和2447个唯一的第一个三位和四位EC编号,以及37 178条具有已知催化位点的链。它还包含与8315个唯一GO项关联的119 004条链。这些数据为I-TASSER服务器中基于结构的功能注释提供了全面的资源。
开发新的留言板系统。
通过电子邮件,信件和电话,我们收到了许多与用户有关我们实验室中开发的服务器系统的问题。为了促进用户之间和/或用户社区与实验室成员之间的交流,在 http://zhanglab.ccmb.med.umich.edu/bbs 建立了讨论板系统。 这有助于在用户之间进行讨论,并在使用程序和/或由我们的实验室成员解释建模结果时快速响应用户的问题。留言板还可以帮助我们收集用户的反馈,这对于帮助我们改善算法和服务以更好地为社区服务至关重要。用户社区目前每天访问董事会的访问量约为3000-4000次。自2011年推出以来,我们已经收到2500多个注册用户的服务评论并回复了1500多个评论/主题,这表明该留言板是提高用户参与度的有效论坛,也是改善服务的绝佳渠道。
三、服务器的输入和输出
3.1 输入
I-TASSER服务器的输入是查询蛋白的主要氨基酸序列。 该服务器提供了三个附加选项,供高级用户输入更多信息以指导I-TASSER建模。
- 第一个选择是指定距离约束和结构模板以辅助建模。
- 第二个是从I-TASSER模板库中排除某些模板,这些模板是出于某些特殊目的而设计的(例如,用于基准测试)。
- 第三个允许用户指定特定残基的二级结构。 对于具有约400个残基的蛋白质,服务器需要10到24小时才能生成完整的建模结果。
3.2 输出量
对于每个提交,分配一个唯一的作业ID和一个URL来跟踪其建模状态。建模完成后,将通过电子邮件通知用户,并在网页上以指定的URL报告结果数据。可以在以下位置获得示例输出页面: http://zhanglab.ccmb.med.umich.edu/I-TASSER/example 。输出数据包括:(i)提交的序列和局部结构特征预测的摘要,(ii)使用的前十个线程模板,(iii)具有全局和局部精度估计的排名最高的3D结构模型,(iv)具有与查询相似的结构的前10种蛋白质,以及(v)配体结合位点,EC号和GO术语的基于结构的功能注释。建模结果在服务器上保存90天,之后将被删除,以节省系统中的磁盘空间。结果页面上列出的所有建模结果都一起收集在一个tarball文件中,该文件可在同一页面上下载。鼓励用户将该文件下载到他们的计算机上以永久存储结果。输出结果在下面简要介绍。在结果注释页面上提供了I-TASSER输出的图形说明:http://zhanglab.ccmb.med.umich.edu/I-TASSER/annotation 。
提交的序列和预测的结构特征。 I-TASSER结果页面的前四部分总结了提交的氨基酸序列和预测的局部结构特征,包括二级结构,溶剂可及性和标准化的B因子,如图2所示。通常,正B-因子值表明残基在结构中更容易被吸收,而负值表明残基相对更稳定。预测的二级结构也显示在B因子图中。位于环或尾部区域的残基往往具有较高的预测B因子值,因为与位于其他规则二级结构区域的残基相比,它们通常较不稳定。提供这些部分时,还会列出用户指定的约束,包括模板对齐和二级结构约束。

3.3 I-TASSER使用的前10个模板
使用LOMETS(20)中当前的14个线程程序集,I-TASSER最多使用140个模板来提取距离约束。但是,由LOMETS排名的前10个模板是最相关的模板,因为它们在约束收集中的权重更高,并在副本交换蒙特卡洛模拟中用作低温副本的起始模型。这些模板的信息在结果页的第f部分中列出,其中包括:(i)模板PDB ID,(ii)标准化 threading Z分数,(iii)alignments覆盖,(iv)sequence identities (v)查询与模板之间的比对。尽管Z得分对应于原始比对得分与标准偏差均值之间的差,但归一化的Z得分定义为Z得分除以程序特定的Z得分隔断。因此,标准化的Z分数> 1表示状态对齐。如果每个线程程序平均至少有一个标准化Z得分> 1的模板,则将查询蛋白归类为“轻松”目标。否则,它被认为是“硬”(Hard)目标。
3.4 I-TASSER预测的前5个模型。
结果页的第六部分报告了多达5个完整的结构模型,以及估计的整体和局部精度。 如果建模模拟相互融合,则报告的模型可能少于5个,这通常表明模型具有相对较高的可信度。 图3显示了示例蛋白质的第一个I-TASSER模型。 它的全局C分数为0.9,估计的TM分数和RMSD分别为0.84和2.4Å。 用户可以将模型的PDB格式的结构文件下载到自己的计算机上,以便在本地可视化该结构。 通过单击网页上的“模型的估计局部精度”链接,也可以下载用于特定残渣的局部精度估计的数据文件和预测的B因子值。

3.5 PDB中的结构类似物 Structure analogs in PDB
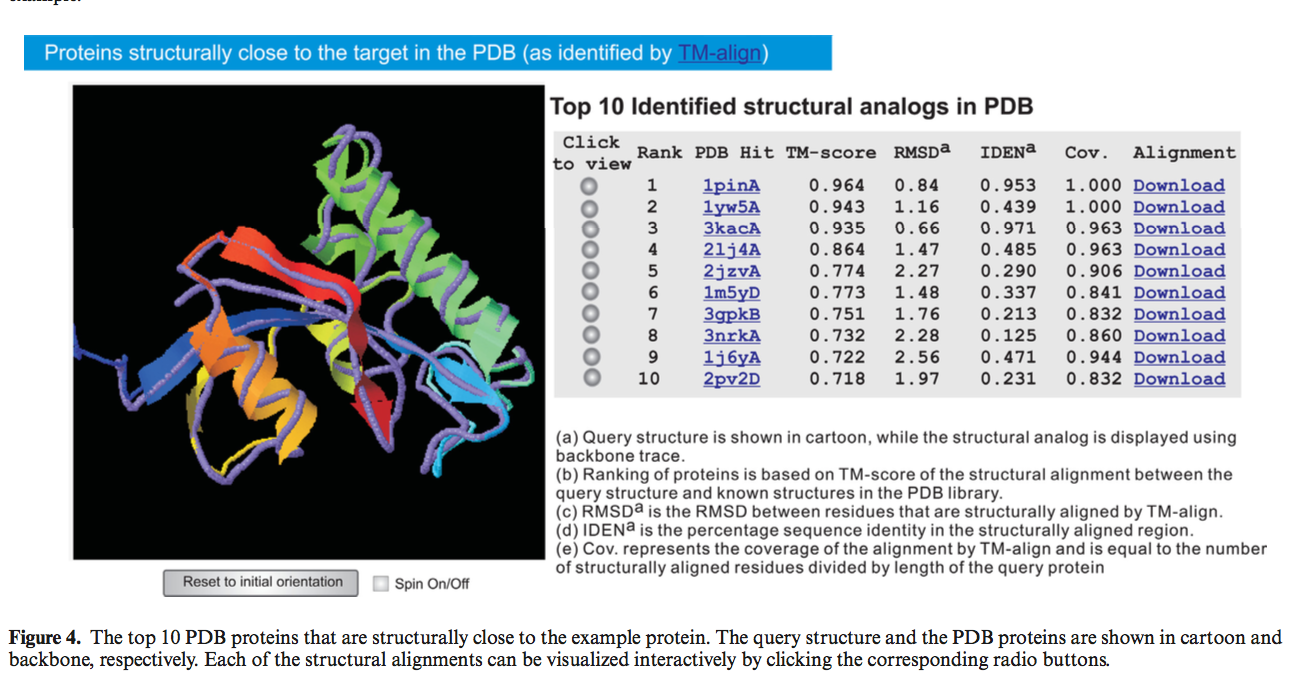
通过TM-align(31)在PDB库中搜索第一个I-TASSER模型,以找到结构类似于查询蛋白的类似物。 图4显示了搜索结果的示例。 查询和10个最接近的蛋白质之间的结构比对通过TM评分(32)进行排名。 该表提供了结构比对的数字细节,包括TM得分,比对覆盖率,RMSD和结构比对区域中的序列同一性。 在同一表中提供了下载叠加结构的坐标文件的链接。 请注意,本节中列出的蛋白质可能与“ I-TASSER使用的十大threading模板”一节中列出的蛋白质不同,因为它们是通过不同方法检测的。 前者是通过基于第一个I-TASSER模型的结构比对检测的,而后者是通过查询序列中的threading发现的。
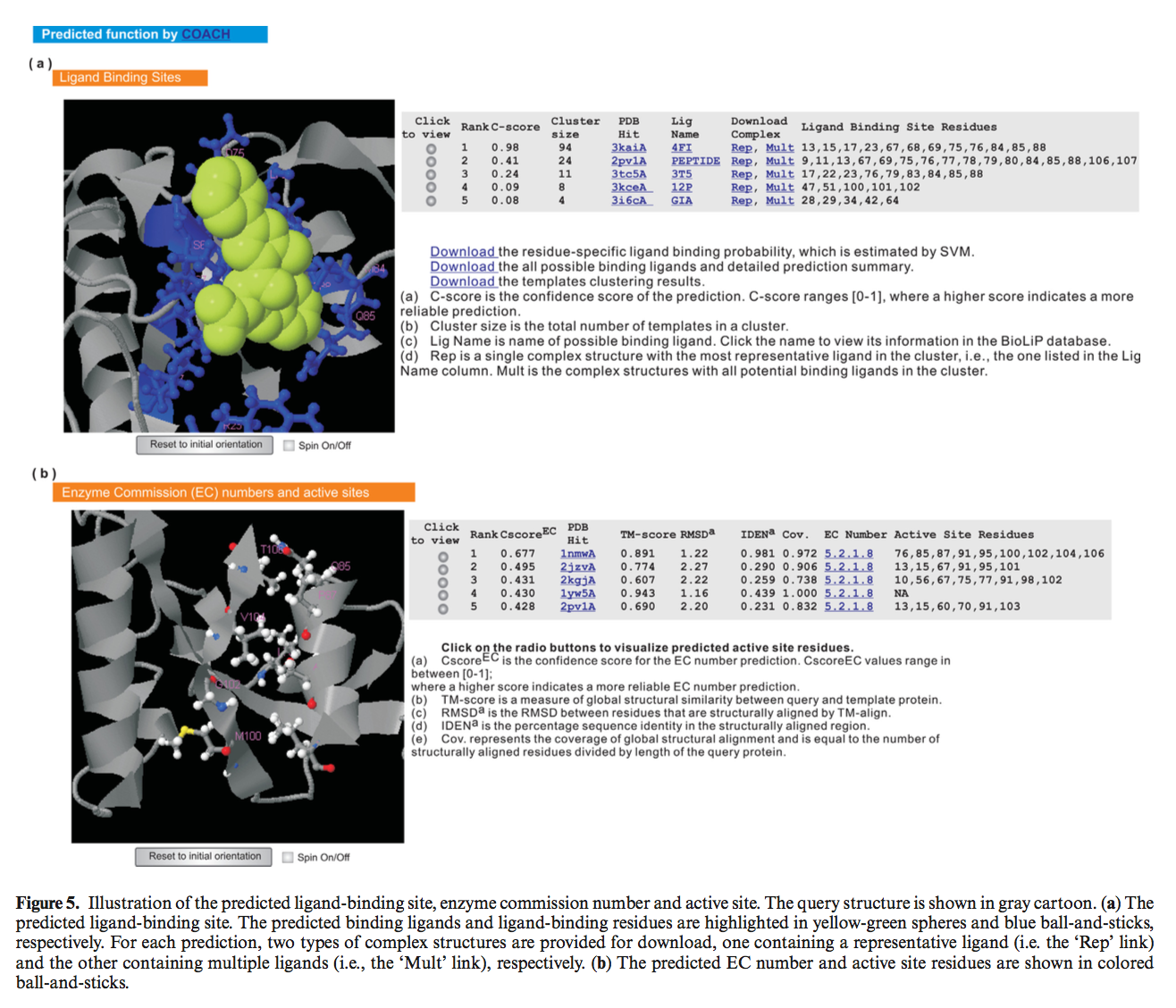
3.6 COACH基于结构的功能注释
通常,具有最高置信度得分的第一个I-TASSER模型已提交给COACH(10),以预测其生物学功能,包括配体结合位点,EC编号和GO术语。 预测的配体结合位点,EC数和活性位点的说明性示例如图5所示。预测的GO术语在结果页面的最后一部分中可用,分为两部分。 第一部分列出了使用GO术语注释的排名前10位的模板蛋白。 由于模板蛋白可能具有其他功能域,因此在最高功能水平上对三个功能方面(分子功能,生物学过程和细胞成分)中最频繁出现的GO术语进行了核对, GO术语在第二部分中介绍。
四、服务器的性能
基于I-TASSER的算法已在基准测试(8,10,23,33)和盲测(26,27,29,34)中进行了广泛的测试。 对于盲法测试,它参加了针对蛋白质结构和功能预测的社区CASP(35)和CAMEO(15)实验。 I-TASSER(组名“ Zhang-Server”)排名第一 在第7届-11日CASP竞赛中自动预测蛋白质结构的服务器(26,27,29,34)。 在CAMEO(15)中,COACH生成了4570个靶标的配体结合位点预测(2012年12月7日至2015年1月9日之间),平均AUC得分为0.84,比第二好的方法高18%。 本实验这些数据表明,I-TASSER服务器代表了自动化蛋白质结构和功能预测的最前沿算法之一。
五、结论
I-TASSER服务器是一种在线工具,用于自动蛋白质结构预测和基于结构的功能注释。所涉及的算法已在社区范围内的盲目实验中进行了严格评估,与蛋白质结构和功能预测中的同类方法相比,具有相当大的优势。在用户社区的大量反馈下,服务器已进行了多种新开发,以改善原子级结构管理,基于结构的功能注释,本地质量估计和用户界面通信中的服务器质量。
用于系统结构和功能预测的服务器系统模板库每周更新一次,可在I-TASSER主页上免费下载。鼓励用户在我们的留言板上报告和讨论I-TASSER服务器相关的问题。我们相信,在用户社区的帮助下以及方法的不断发展,I-TASSER服务器正朝着基于最新方法提供最准确,最有用的结构和功能预测的目标迈进。
参考资料
- I-TASSER server: new development for protein structure and function predictions
