【5.2.2】nanopolish-polya
由于均聚物效应(homopolymer)和与测序接头的接近,从纳米孔天然 RNA 测序中获得的读数的多聚腺苷酸尾被不正确地碱基命名(basecalled),使其长度难以量化。我们开发了polya子程序以使用替代统计模型来估计这些尾长。
在本快速入门教程中,我们将向您展示如何从原始 fast5 文件开始逐步估算多聚腺苷酸化尾长。我们将使用 Oxford Nanopore Technologies 的 albacore basecaller 对 fast5 文件进行碱基调用,然后将结果读取与minimap2对齐,使用nanopolish index索引文件,最后分割读取并使用nanopolish polya调用尾长。
一、分析流程
1.callbase
mkdir data && mkdir data/fastqs
wget ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR276/ERR2764784/30xpolyA.tar.gz -O 30xpolyA.tar.gz && mv 30xpolyA.tar.gz data/
tar -xzf data/30xpolyA.tar.gz -C data/
guppy_basecaller -c rna_r9.4.1_70bps_hac.cfg -i data/30xpolyA/fast5/pass -s data/fastqs
cat data/fastqs/*.fastq > data/30xpolyA.fastq
注:
-f and -k arguments based on your flow-cell and sequencing kit, as the basecaller’s accuracy is highly dependent upon these settings
2.建索引
nanopolish index --directory=data/30xpolyA/fast5/pass --sequencing-summary=data/fastqs/sequencing_summary.txt data/30xpolyA.fastq
3.比对
wget https://raw.githubusercontent.com/paultsw/polya_analysis/master/data/references/enolase_reference.fas
wget https://raw.githubusercontent.com/paultsw/polya_analysis/master/data/references/enolase_reference.fas.fai
mv enolase_reference.* data/
minimap2 -a -x map-ont data/enolase_reference.fas data/30xpolyA.fastq | samtools view -b - -o data/30xpolyA.bam
cd data && samtools sort -T tmp -o 30xpolyA.sorted.bam 30xpolyA.bam && samtools index 30xpolyA.sorted.bam && cd ..
4.评估polya
nanopolish polya --threads=8 --reads=data/30xpolyA.fastq --bam=data/30xpolyA.sorted.bam --genome=data/enolase_reference.fas > polya_results.tsv
二、结果解读
head -20 polya_results.tsv | column -t
readname contig position leader_start adapter_start polya_start transcript_start read_rate polya_length qc_tag
d6f42b79-90c6-4edd-8c8f-8a7ce0ac6ecb YHR174W 0 54.0 3446.0 7216.0 8211.0 130.96 38.22 PASS
453f3f3e-d22f-4d9c-81a6-8576e23390ed YHR174W 0 228.0 5542.0 10298.0 11046.0 130.96 27.48 PASS
e02d9858-0c04-4d86-8dba-18a47d9ac005 YHR174W 0 221.0 1812.0 7715.0 8775.0 97.16 29.16 PASS
b588dee2-2c5b-410c-91e1-fe8140f4f837 YHR174W 0 22.0 8338.0 13432.0 14294.0 130.96 32.43 PASS
每行对应于给定读取的输出。这些列具有以下解释:
-
readname是指与此特定读取关联的唯一 ID。该字符串还用于查找相应的 fast5 文件,例如通过查找通过nanopolish index生成的readdb文件。
-
contig是指此读取对齐的参考序列,并取自 BAM 文件。
-
position是与参考序列比对的 5' 起始位置,也来自 BAM 文件。
-
leader_start、adapter_start、polya_start和transcript_start是nanopolish中底层模型生成的信号分割的索引 。简而言之,每个 fast5 文件中的电流读数原始序列有四个具有生物学意义的区域;这四个条目表示每个连续区域的起始索引。索引从 0 开始,朝向 3' 到 5' 方向(由于天然 RNA 纳米孔测序协议的固有方向)。补充说明< https://www.biorxiv.org/content/biorxiv/suppl/2018/11/09/459529.DC1/459529-2.pdf 中提供了此分割算法的完整 说明到我们的相关出版物。
-
read_rate是通过孔读取的估计易位(translocation)速率(以核苷酸/秒为单位)。即使是单个分子的测序过程中,易位率也是不均匀的,因此这最终是动态的、时变率的汇总统计。
-
polya_length是估计的多聚腺苷酸化尾长,以核苷酸数计。这个值是浮点数而不是整数反映了这样一个事实,即我们估计的尾长是基于易位率(translocation)的估计器的输出。
-
qc_tag是一个附加标志,用于指示估计的有效性。一般来说,您应该只使用此值设置为PASS的输出文件的行;所有其他(例如)将qc_tag设置为SUFFCLIP、ADAPTER等的行都显示出不规则的迹象,表明我们认为估计是不可靠的。您可以使用grep轻松过滤掉所有标签设置为PASS以外的任何行:
grep ‘PASS’ polya_results.tsv > polya_results.pass_only.tsv
三、算法理解
- 信号区域划分
- 依据polya区域的时间,推测A的长度
Biologically, each sequenced read consists of a sequencing adapter (which we call the leader region), the RT splint adapter (which we call the adapter region), the polyadenylated tail, and the coding transcript, respectively, from 3’ to 5’
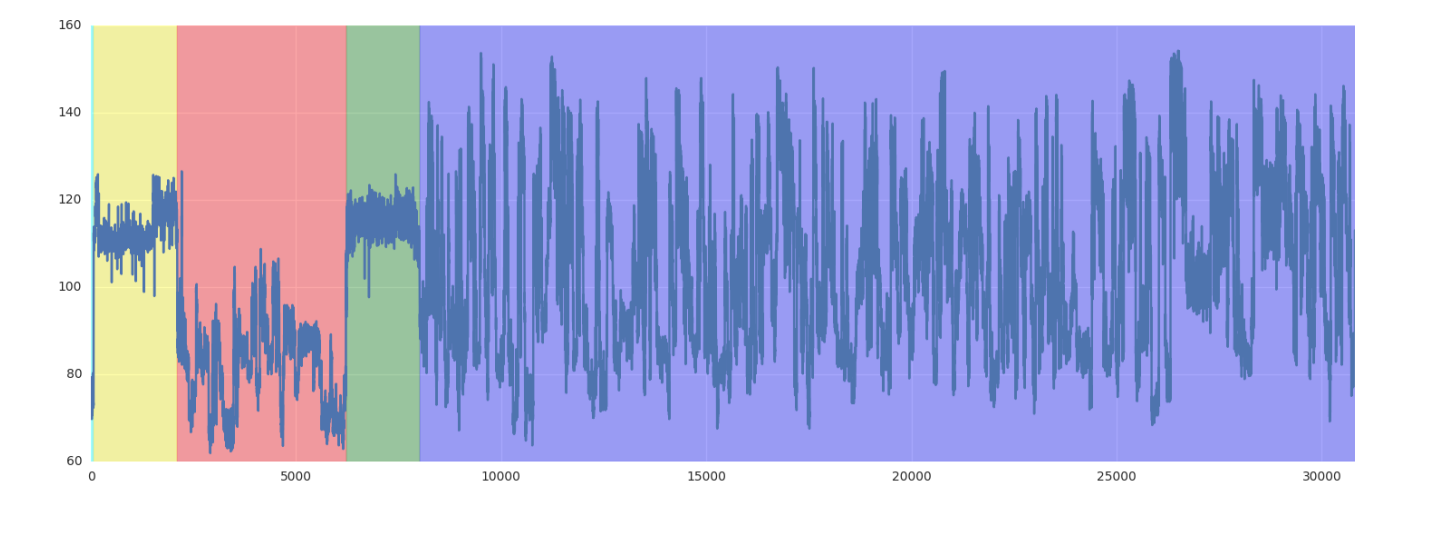
Figure 1: An example of a squiggle segmentation generated by the hidden markov model. Distinct regions, from left to right: start (cyan), leader (yellow), adapter (red), poly(A) tail (green), and transcript (purple). Two samples flagged as “cliffs” can be observed in the poly(A) tail.
参考资料
