【2.9.1】AMBER分子动力学简例(amber8?)
通过这个教程,大概知道了amber动力学分析的流程。因为这个版本比较旧了,不建议看这个版本的学习。
AMBER分子动力学程序包是加州圣弗兰西斯科大学(University of California San Francisco,UCSF)的Peter A Kollman和其同事编的,程序很全,现在已经发展到版本9.0。AMBER功能涵盖种类非常多的生物分子,包括蛋白、核算以及药物小分子。软件详细情况请浏览http://amber.scripps.edu.
一、基本概念
以下是AMBER软件包中四个主要的大程序:
-
Leap:用于准备分子系统坐标和参数文件,有两个程序:
-
xleap:X-windows版本的leap,带GUI图形界面。
-
tleap:文本界面的Leap。
-
Antechamber:用于生成少见小分子力学参数文件的。有的时候一些小分子Leap程序不认识,需要加载其力学参数,这些力学参数文件就要antechamber生成。
-
Sander:MD数据产生程序,即MD模拟程序,被称做AMBER的大脑程序。
-
Ptraj:MD模拟轨迹分析程序。
二、学习案例
本教程研究的题目是脑下垂体荷尔蒙之一的oxytocin,需要X光衍射晶体结构文件1NPO.PDB。该文件包含了该荷尔蒙和其运载蛋白的复合物,可以从蛋白数据库下载。
PDB文件是不包含氢原子的,Leap程序会自动的加上PDB文件缺少的东西。当第一次使用PDB文件的时候,要十分留意文件包含的信息,所以PDB文件缺少的残疾、侧链或者添加的变异都在这个地方记录。可以用文本阅读程序阅读PDB文件头部的信息,即以REMARKS开头的信息文本行。PDB一个重要的信息是SSBOND记录,该记录说明结构中二硫键的位置,这样的信息在使用Leap程序建立分子结构的时候需要。在本教程中,我们将比较oxytocin在真空和溶液中分子动力学的差异,如果没有二硫键,将影响整个结果
2.1 准备输入文件 Prepare Input File
AMBER套件包含60多个程序。 但是,要进行传统的分子动力学模拟,您只需要知道两个程序:tleap和sander。 除了这两个之外,您还可能会使用其他两个帮助程序carnal和ptraj。 这两个程序是AMBER套件的数据分析主力,但是如果您知道其他可以使用AMBER轨迹文件的分析程序,也可以改用它们。
像计算机上的其他所有内容一样,运行计算机模拟完全是关于输入和输出的。 sander是整个套件的主要simulation引擎;它需要两个输入文件来描述要模拟的分子系统,一个控制文件指定要模拟的条件,并根据这些信息来计算经典的分子动力学轨迹。 tleap是帮助程序,它使用预定的坐标文件(例如pdb)并生成拓扑文件和重新启动文件。当然,我在这里简化了一点。 tleap实际上不仅仅可以做到这一点,而且它不是AMBER中唯一可以获取pdb文件并为sander生成输入文件的程序。另一个帮助程序称为xleap。它们之间的区别在于xleap使用图形界面,而tleap使用基于文本的界面。刚开始使用AMBER时,我紧紧抓住图形的xleap并拒绝“后退”的基于文本的tleap。随着时间的流逝,我逐渐认识到基于文本的界面的价值。如果您打算进行很多模拟,则需要花费一些额外的精力来习惯基于文本的界面。
整合酶核心域(pdb id 1qs4)的最新x射线结构具有三个晶体学单体。在环区域中也缺少一些残基。在将pdb文件放入tleap之前,我们必须首先解决这些问题。如何对pdb文件中的遗留残基进行建模在很大程度上取决于您的研究目的以及所使用的特定模型。如果您只是刚开始建模而又不知道该怎么做,南缅因大学的Gale Rhodes教授可以使用免费提供的SwissPDB Viewer程序( http://www.usm.maine.edu/~rhodes/SPVTut/ )。在这项研究中,我使用了整合酶的另一种晶体结构(pdb id 1bis)对缺失的残基进行了建模。除了缺失的残基外,在133和185位还有两个点突变。引入这些突变主要是为了帮助蛋白质更好地形成晶体。但是,后来认为F185K突变会影响附近第二个表面环的动力学。由于我们对表面回路的动力学感兴趣,因此应将其从Lys更改回Phe。 E133W突变不在蛋白质的任何关键区域,pdb文件实际上表明pdb序列和相应的序列数据库之间存在一些冲突,因此我们可以不理会晶体坐标。如今,计算机中的残基突变比试管中的突变更容易。您可以使用许多免费的图形化分子编辑程序。我使用SwissPDB Viewer这样做是因为它是免费的,而且还因为它具有内置的能耗最小化功能来稍微清理建模的结构。为了节省您的时间,可以从“下载”页面下载清理后的结构,或单击“此处”。
下载文件:
wget -c https://ambermd.org/tutorials/advanced/tutorial8/files/mg.off
wget -c https://ambermd.org/tutorials/advanced/tutorial8/files/wt1mg.pdb
下载文件后,请浏览内部。 您会注意到没有氢原子。 如果您不想使用我准备的文件,但是想要准备自己的pdb文件,请确保立即剥离所有氢原子。 根据用于构建初始模型的软件的不同,最终可能会产生不兼容的氢名称。 仅仅将它们剥离并让氢添加就更容易了。
在命令行上,启动tleap程序。 您应该会看到以下内容:
$ tleap
-I: Adding /amber/dat/leap/prep to search path.
-I: Adding /amber/dat/leap/lib to search path.
-I: Adding /amber/dat/leap/parm to search path.
-I: Adding /amber/dat/leap/cmd to search path.
Welcome to LEaP!
Sourcing leaprc: /amber/dat/leap/cmd/leaprc
Log file: ./leap.log
Loading parameters: /amber/dat/leap/parm/parmME.dat
Loading library: /amber/dat/leap/lib/all_nucleic94.lib
Loading library: /amber/dat/leap/lib/all_aminoME.lib
Loading library: /amber/dat/leap/lib/all_aminoctME.lib
Loading library: /amber/dat/leap/lib/all_aminontME.lib
Loading library: /amber/dat/leap/lib/ions94.lib
Loading library: /amber/dat/leap/lib/water.lib
>
不用担心目录中的差异。 可以使用您自己的jumprc文件来指定。 稍后,您可以了解如何定义您的jumprc文件。 现在,只接受默认值即可。
如果您在启动tleap时遇到问题,请检查是否正确设置了环境变量。 如果您使用的是bash shell,请确保〜/ .bash_profile中包含这两行
export AMBERHOME=/amber
export PATH=$PATH:/amber/exe
If you are a C-Shell user, then add the following to your ~/.cshrc file.
setenv AMBERHOME "/amber"
setenv PATH "${PATH}:/amber/exe"
where /amber is the location of your AMBER distribution.
接下来,我们将pdb文件加载到tleap中,并将其分配给我们称为“ mol”的变量
...
...
Loading library: /amber/dat/leap/lib/water.lib
> mol = loadpdb wt1mg.pdb
Loading PDB file: ./wt1mg.pdb
Unknown residue: MG number: 154 type: Terminal/last
..relaxing end constraints to try for a dbase match
-no luck
Added missing heavy atom: .R<CGLN 154>.A<OXT 18>
Creating new UNIT for residue: MG sequence: 155
Created a new atom named: MG within residue: .R<MG 155>
total atoms in file: 1189
Leap added 1192 missing atoms according to residue templates:
1 Heavy
1191 H / lone pairs
The file contained 1 atoms not in residue templates
>
如果一切正常,则不应从tleap收到任何错误消息。 通常,您第一次尝试时可能会收到某种错误消息。 像这里一样,tleap抱怨有一个未知的残基MG。 这是因为启动程序时加载的参数库中未包含MG。 如果您的本地AMBER安装中已经包含MG参数,那么您应该不会看到此问题。 如果不是,则可以从“下载”页面下载参数文件的副本,。 退出tleap,将下载的文件放置在当前工作目录中,再次启动tleap,然后发出命令“ loadoff MG.off”,然后再次加载loadpdb。 屏幕现在应如下所示:
Welcome to LEaP!
Sourcing leaprc: /amber/dat/leap/cmd/leaprc
Log file: ./leap.log
Loading parameters: /amber/dat/leap/parm/parmME.dat
Loading library: /amber/dat/leap/lib/all_nucleic94.lib
Loading library: /amber/dat/leap/lib/all_aminoME.lib
Loading library: /amber/dat/leap/lib/all_aminoctME.lib
Loading library: /amber/dat/leap/lib/all_aminontME.lib
Loading library: /amber/dat/leap/lib/ions94.lib
Loading library: /amber/dat/leap/lib/water.lib
> loadoff MG.off
Loading library: ./MG.off
> mol = loadpdb wt1mg.pdb
Loading PDB file: ./wt1mg.pdb
Added missing heavy atom: .R<CGLN 154>.A<OXT 18>
total atoms in file: 1189
Leap added 1192 missing atoms according to residue templates:
1 Heavy
1191 H / lone pairs
>
tleap程序足够聪明,可以识别C端缺少OXT原子,因此会自动为我们添加。 这不是错误消息。
至此,我们已经加载了pdb文件,添加了氢和缺少的重原子,并为所有原子分配了参数。 下一步将需要大量的计算。 我们将添加一个水箱并向系统添加抗衡离子以中和电荷。
实际上,这里并没有引入立场,正确的流程是这样的:
tleap -s -f /data/software/amber/amber18/dat/leap/cmd/oldff/leaprc.ff99SB
loadoff mg.off
mol = loadpdb wt1mg.pdb
f后面接的是力场
当然也可以通过在tleap界面中 source leaprc.ff03 来添加力场
2.1.2 用tleap添加水和离子
现在,我们将使用tleap将水箱添加到分子和抗衡离子中,以完成模型系统。
> solvateBox mol WATBOX216 10
Solute vdw bounding box: 49.995 53.684 37.063
Total bounding box for atom centers: 69.995 73.684 57.063
Solvent unit box: 18.774 18.774 18.774
Total vdw box size: 73.439 76.851 60.149 angstroms.
Volume: 339472.593 A^3
Total mass 162173.797 amu, Density 0.793 g/cc
Added 8064 residues.
>
如上所示,添加水箱的命令称为“ solvateBox”。 还有其他几种加水的方法,但我们这里只坚持最简单的方法。 WATERBOX216是TIP3P水的预平衡盒。 数字10是盒子边缘和蛋白质之间的缓冲距离(以埃为单位)。 在这里,您必须对要使用的缓冲区大小做出一些判断。 如果您使用的数字太大,最终会得到一个大的水箱,并在没必要的水分子上浪费了很多不必要的计算时间。 但是,如果您使用的水箱太小,则在模拟过程中,分子可能会发生构象变化,并且部分分子可能会粘在水箱外部。 如果您想在接近实验条件的条件下进行模拟并且知道系统的浓度,则可以使用该信息来计算出所需水箱的大小并进行明确设置,否则,我认为10是一个合理的数字 。
solvatebox添加的水环境是一个立方体,不是八面体。一般要求水表面到蛋白距离要达到8.5纳米,避免MD过程中蛋白冲出水环境,但是水环境越大计算时间将越长。如果MD过程内含PME,那么水箱长度要大于2倍截矩(cutoff),截矩在MD配置文件中定义。
这个地方,我是报错了
> > solvateBox mol WATBOX216 10
> solvateBox: Argument #2 is type String must be of type: [unit]
> usage: solvateBox <solute> <solvent> <buffer> [iso] [closeness]
通过List可以看到所有的units,
> list
ACE ALA ARG ASH ASN ASP CALA CARG
CASN CASP CCYS CCYX CGLN CGLU CGLY CHCL3BOX
CHID CHIE CHIP CHIS CILE CIO CLEU CLYS
CMET CPHE CPRO CSER CTHR CTRP CTYR CVAL
CYM CYS CYX Cl- Cs+
TIP3PBOX
所以,是这个立场,没有这个水盒子,改成了
solvateBox mol TIP3PBOX 10
我们需要做的下一件事是添加平和离子(counter ions)。 在发出“加法”命令之前,我们需要确定系统是带正电荷还是带负电荷。 如果带正电荷,我们将要添加带负电荷的Cl-来抵消它,如果带负电荷,那么我们将添加Na+来抵消它。 要计算系统的费用,我们可以使用以下命令“ charge”:
> charge mol
Total unperturbed charge: 2.00
Total perturbed charge: 2.00
>
因此,我们看到我们的系统带正电荷。我们将添加Cl- 原子以平衡电荷。
AMBER实际上提供了两种添加离子的算法。
- “addions”中实现的第一种方法是简单地在溶质周围绘制一个网格并将离子放置在能量最低的网格点上。这种方法将忽略水分子在放置离子的位置,如果所选位置与水分子重叠,则会删除水并将其替换为离子。如果使用这种算法,我们最终会在我们的 Mg2+ 旁边得到 Cl- 离子,这不是我们想要的。
- 由命令“ addions2”实现的第二种方法与“ addions”几乎具有相同的功能,除了它将溶剂分子与溶质相同。我们将使用“ addions2”来确保Cl-离子与我们的分子相距一定距离,因此其电荷不会人为地扭曲我们的系统。命令末尾的数字“ 0”表示我们希望回避以找出适当数量的抗衡离子以中和整个系统。
如您所见,此计算在我的Pentium IV PC上花费了990秒,因此,如果您的计算机在一段时间内似乎什么都没有发生,请稍等一点耐心,然后再单击“重置”按钮。
> addIons2 mol Cl- 0
2 Cl- ions required to neutralize.
Adding 2 counter ions to "mol" using 1A grid
Grid extends from solute vdw + 2.47 to 8.47
Resolution: 1.00 Angstrom.
grid build: 4 sec
Calculating grid charges
charges: 990 sec
Placed Cl- in mol at (-18.42, 1.87, 29.98).
Placed Cl- in mol at (0.58, -17.13, 29.98).
Done adding ions.
>
最后,我们准备输出结果并将系统保存为输入文件。
> saveAmberParm mol wt1mg.parm7 wt1mg.crd
Checking Unit.
Building topology.
Building atom parameters.
Building bond parameters.
Building angle parameters.
Building proper torsion parameters.
Building improper torsion parameters.
total 468 improper torsions applied
Building H-Bond parameters.
Marking per-residue atom chain types.
(Residues lacking connect0/connect1 -
these don't have chain types marked:
res total affected
CGLN 1
NCYS 1
WAT 8064
)
(no restraints)
>
输入 quit 退出tleap
2.2 能量优化
创建初始模型后,最好以肉眼检查模型以确保一切看起来合理。 您可以使用命令“ ambpdb”将拓扑转换并重新启动文件为pdb格式:
$ ambpdb -p wt1mg.parm7 < wt1mg.crd > wt1mg_solvated.pdb
| New format PARM file being parsed.
| Version = 1.000 Date = 06/14/03 Time = 14:53:33
$
然后,您可以使用喜欢的可视化软件检查pdb文件。 同样,我在这里以SwissPDB Viewer为例。 将pdb文件加载到查看器中时,应该会看到类似的内容。

在SwissPDB Viewer中,“选择”菜单下有一个名为“ aa making clashes”的选项。 此命令将突出显示与其邻居接触不良的氨基酸残基。 要显示冲突,请从“颜色”菜单中选择“按选择”。 您现在应该看到类似以下的内容:

蓝色区域表示接触不良的残基。 还不错,只有三个。
在开始分子动力学模拟之前,我们需要消除这些不良接触。 原因是,如果我们从这些不良接触开始分子动力学,则该区域中的能量将非常高,并且可能使模拟崩溃或导致轨迹朝不现实的方向前进。
我们通过最小化能量来消除这些不良接触。 即使没有明显的不良接触,还是要进行短暂的能量最小化以稍微放松结构,这仍然是一个好主意。
我们将分两个阶段进行能量最小化。 在第一阶段,我们将仅使水分子最小化,并保持蛋白质和Mg2+固定。 以下是我用于此计算的控制输入文件:
Minimization with Cartesian restraints for the solute
&cntrl
imin=1, maxcyc=200,
ntpr=5,
ntr=1,
&end
Group input for restrained atoms
100.0
RES 1 155
END
END
将这些控制信息保存在一个名为“ min.in”的文件中,将其像这样输入到sander:
$ sander -O -i min.in -p wt1mg.parm7 -c wt1mg.crd -r wt1mg_min.rst -o wt1mg_min_water.out -ref wt1mg.rst
注意命令行末尾的“ -ref”选项。 通常不需要此选项。 在这种情况下,我们需要使用它,因为我们正在进行受限的最小化,并且此信息对于sander来说是必需的,以便能够映射我们在“ min.in”中指定的残渣选择。 文件“ wt1mg.rst”与在tleap准备结束时生成的原始重新启动文件相同。 只需将原始文件wt1mg.crd复制到wt1mg.rst。 (我们制作同一文件的另一个副本的原因仅是为了记账。您可以根据需要使用相同的wt1mg.crd。)
此命令要求sander将“ min.in”作为控制输入,将wt1mg.parm7作为系统的参数文件,将wt1mg.crd作为输入坐标。 它将写出wt1mg.rst作为重新启动文件,并写出wt1mg_min_water.out作为输出文件。

计算可能需要几分钟才能完成,因此这是短暂休息的好时机。
先前的最小化计算使溶质分子周围的水松弛。 如果我们想非常细致,现在可以进行相反的操作,并在保持水分子固定的同时仅使溶质最小化,然后将整个系统放在一起。 但是,请记住,我们的目标只是消除不良的联系,因此不必过度考虑最小化。 我们将直接着手最小化整个系统:
Minimization of the entire molecular system
&cntrl
imin=1, maxcyc=200,
ntpr=5,
&end
同样,将此输入保存在文件中,并命名为“ min_all.in”,然后将其输入到sander。
旁注:这提醒我告诉您,在执行模拟时,文件数量可以快速累加。 如果您习惯养成一个单独的笔记本来跟踪事物,那很好,但是如果您像我一样并且很少在纸上写下任何东西,那么拥有一个好的文件命名系统非常重要。 。 拥有一个好的命名系统可以使您的工作或多或少地自我记录,而拥有一个糟糕的命名系统可能会在接下来的几周内造成很多不必要的麻烦。
$ sander -O -i min_all.in -p wt1mg.parm7 -c wt1mg_min_water.rst -r wt1mg_min_all.rst -o wt1mg_min_all.out
在快速的Pentium IV机器上,这将再花费5到10分钟,因此该是另一个短暂的休息时间了。
当您回来时,不要忘记像以前一样通过从重新启动文件生成pdb文件来检查结果。
2.3 平衡 Equilibration
在MD模拟中,大分子和周围溶剂的原子经历弛豫(relaxation),通常在系统达到稳态之前会持续数十或数百皮秒(picoseconds)。模拟轨迹的初始非平稳段通常在平衡特性的计算中被丢弃。 MD模拟的这一阶段称为平衡阶段(equilibration stage.)。
平衡方案在很大程度上仍然取决于个人喜好。一些流程要求非常复杂的过程,包括以逐步的方式逐渐提高温度,而其他更具侵略性的方法只是使用线性温度梯度并将系统加热到所需温度。
在我们的示例中,我们将遵循AMBER手册中建议的协议,并执行两阶段平衡。在第一阶段,我们将从100 K的低温启动系统,并在10皮秒的simulation时间内逐渐加热至300K。我们将在保持体积不变的情况下执行此平衡阶段。输入文件如下:
Heating up the system equilibration stage 1
&cntrl
nstlim=5000, dt=0.002, ntx=1, irest=0, ntpr=500, ntwr=5000, ntwx=5000,
tempi =100.0, temp0=300.0, ntt=1, tautp=2.0, ig=209858,
ntb=1, ntp=0,
ntc=2, ntf=2,
nrespa=2,
&end
第1行: 我们指定要运行5000步(nstlim = 5000),时间步长为2 fs(dt = 0.002);我们正在开始新的运行,因此没有以前的速度信息(ntx = 1,irest = 0),我们将每5000步打印一次能量输出(ntwr = 5000),并每5000个坐标保存一次坐标(ntwx = 5000)。 第2行: 我们希望使用Berendsen耦合算法将初始温度设为100K(tempi = 100.0),将参考温度设为300K(temp0 = 300.0),以保持恒定温度。温度耦合的时间常数为2 ps(tautp = 2.0)。对于初始速度分配,需要一个随机数种子(ig = 209858)。如果使用相同的种子,您将获得完全相同的轨迹。如果要以不同的初始速度分配运行多个轨迹,则需要使用不同的随机种子编号。
第3行: 我们将使用体积恒定(ntb = 1)且没有压力控制(ntp = 0)的周期性边界
第4行: 我们将使用SHAKE算法来仅约束涉及氢的键(ntc = 2),并忽略这些键的力评估(ntf = 2)。我们还通过重置坐标设置了更严格的公差
第5行: 为了稍微加快计算速度,我们还采用了较大的时间步长(nrespa = 2)来评估力场中的慢变项。在此设置下,时间步长等于nrespa * dt(2 * 2 fs = 4 fs。)
好的,将此输入文件另存为“ eq_v.in”。 现在,让我们启动引擎!
注意:nrespa选项仅适用于AMBER 7及更高版本。如果您使用的是旧版本的AMBER,请记住在输入中排除nrespa。
$ sander -O -i eq_v.in -p wt1mg.parm7 -c wt1mg_min_all.rst -r wt1mg_eq_v.rst -x wt1mg_eq_v.crd -o wt1mg_eq_v.out
在双CPU Pentium III上,此计算花费了2个小时,因此您可以在这里休息一下。 或者,您可以从下载页面下载我的结果。
计算完成后,您的目录中应该还有几个文件。 让我们看一下这些结果。
- wt1mg_eq_v.rst 用于下一次计算的重启文件
- wt1mg_eq_v.crd 保存的快照
- wt1mg_eq_v.out 输出文件,其中包含各种能量项以及温度以及其他内容。
回顾我们这部分平衡的目标是将系统温度从100K升高到300K。 为了检查结果,让我们从输出文件中提取温度信息并将其绘制在图形上。
在命令行上,执行以下操作:
$ grep TEMP wt1mg_eq_v.out | awk '{print $6, $9}' > temp.dat
这将从输出文件中提取时间和温度信息,并将它们保存在名为temp.dat的新文件中。 然后,您可以在自己喜欢的图形或电子表格程序中绘制此数据。 我使用Excel生成以下图形。 请记住要排除最后两个数据项,因为它们是平均值和均方根偏差,而不是实际数据点。
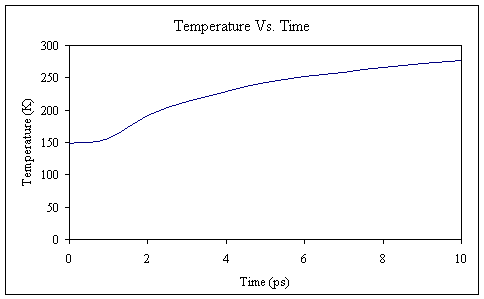
从该图可以看出,在10 ps仿真时间内,系统温度逐渐升高。 它还不是30万,但就我们的目的而言,它已经足够接近了。
接下来,我们将使用压力和温度控制来平衡系统,以将水的密度调整为实验值。
输入文件如下:
Constant pressure constant temperature equilibration stage 2
&cntrl
nstlim=5000, dt=0.002, ntx=5, irest=1, ntpr=500, ntwr=5000, ntwx=5000,
temp0=300.0, ntt=1, tautp=2.0,
ntb=2, ntp=1,
ntc=2, ntf=2,
nrespa=1,
&end
请注意,除了突出显示的项目外,新输入文件与上一个几乎完全相同。 第一个更改是为了反映一个事实,即我们现在从先前的模拟继续进行,并且将使用重新启动文件中的速度信息(ntx = 5,irest = 1)。 我们还通过各向同性位置缩放(isotropic position scaling)(ntp = 1)打开恒定压力(ntb = 2),并摆脱了未使用的参数(tempi和ig)。 对于恒定压力的simulatoins,我们要关闭respa选项。
让我们继续乐趣:
$ sander -O -i eq_pt.in -p wt1mg.parm7 -c wt1mg_eq_v.rst -r wt1mg_eq_pt.rst -x wt1mg_eq_pt.crd -o wt1mg_eq_pt.out
到现在为止,我相信您已经开始掌握它了。 再次,我们将系统平衡为10 ps,并从计算中获得了几个新的输出文件。 此时,我们的系统应具有约300 K的温度和约1克/毫升的密度。
再次,让我们从输出文件中提取温度和密度数据并绘制它们。
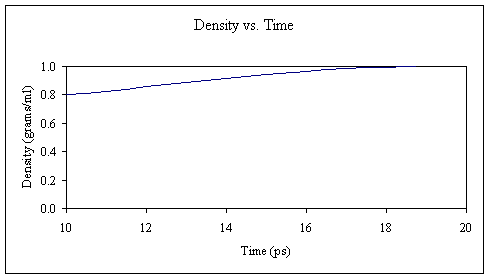
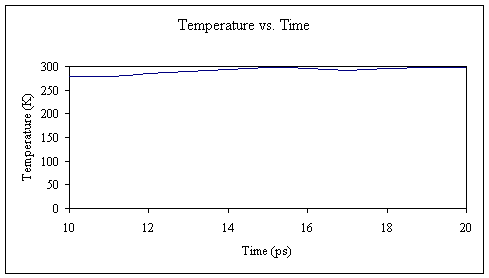
我们已将系统安全地从100 K提升到300 K,并将水箱的密度从〜0.8调整为〜1.0克/毫升。 可以肯定的是,我们将延长恒温和恒压平衡时间稍长一点:
Constant pressure constant temperature equilibration stage 3
&cntrl
nstlim=50000, dt=0.002, ntx=5, irest=1, ntpr=500, ntwr=5000, ntwx=5000,
temp0=300.0, ntt=1, tautp=2.0,
ntb=2, ntp=1,
ntc=2, ntf=2,
nrespa=1,
&end
好吧,所以我低估了。 现在,我们将执行100 ps的平衡运行。 从现在开始,除非您拥有一台非常快的PC或访问超级计算机,否则您可能不想坐在那里等待结果。 但是,您仍然可以进行设置计算的过程,并让计算运行一会儿,以确保一切正确。 然后,我将为您提供结果,以便您可以继续进行下一步。
按照前面的示例,我从输出文件中提取了温度,并将它们组合成一个连续的图。 我们可以看到,在最初的20 ps平衡后,温度肯定已达到所需的300K。 对于其余的100 ps平衡,温度保持相当恒定。
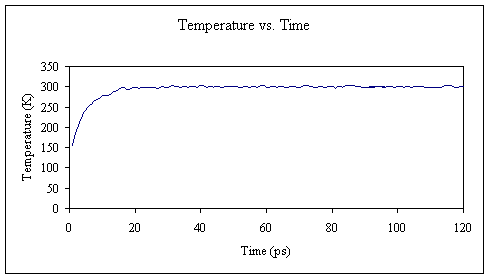
在MD的初始阶段,如果分子的构象不处于固定相(stationary phase),则势能会漂移,因此通常将势能用作确定生产数据之前需要多少平衡的标准。。有助于确定的另一项措施是针对初始结构的RMSD。 由于可以像输出温度值一样直接从输出文件中提取势能值,因此我将继续向您展示结果。
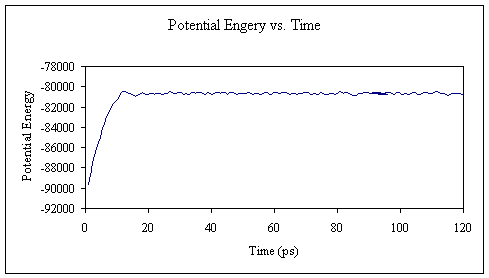
接下来,我将向您展示如何使用程序ptraj执行RMSD计算。
2.3.2 数据分析
数据分析是任何分子动力学模拟项目中最不标准的部分。 通常,人们最终不得不编写自己的程序来以自己想要的方式分析轨迹。 但是对于轨迹的标准处理,carnal和ptraj可以胜任处理此类需求的能力。 在本节中,我们将介绍ptraj的一些常见用法。
1.固定AMBER轨迹(trajectory):
至此,我们已经收集了大约100 ps的MD轨迹。 您现在可以使用UIUC中的VMD程序来可视化轨迹。 但是,当您播放电影时,您会注意到水箱在一段时间后散开。 通过使用ptraj重新成像轨迹可以轻松解决此问题
初始:

最后:

重新成像轨迹输入:
trajin wt1mg_eq.crd
center :1-154
image center familiar
rms first out wt1mg_eq_rms.out :3-152@CA
trajout wt1mg_eq_nice.crd nobox
保存此输入并将其命名为“ ptraj.in”,然后按如下所示运行ptraj:
$ ptraj wt1mg.parm7 ptraj.in
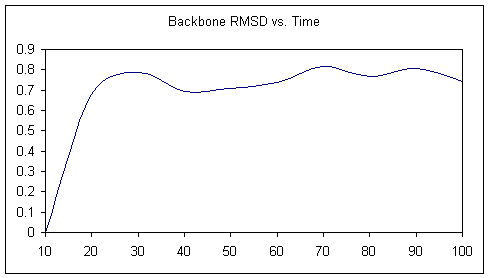
基本上,这将导致ptraj读取由sander创建的原始轨迹文件,并将坐标更新到蛋白质质量中心,然后重新对周期框进行成像,以使其看起来美观而方正。 最后,它将使用第一个帧作为参考来计算每个帧的均方根值,并叠加到C-alpha原子上,从每个末端排除两个残基。 然后将生成的叠加帧写到名为wt1mg_eq_nice.crd的新轨迹文件中,在每个帧的末尾删除框信息。
在屏幕上,您应该看到类似以下内容:
....
....
....
Processing AMBER trajectory file wt1mg_eq.crd
Set 1 ..........
PTRAJ: Successfully read in 10 sets and processed 10 sets.
Dumping accumulated results (if any)
PTRAJ RMS: dumping RMSd vs time data
此处显示的RMSD图表明,骨架构象在约20 ps后已达到稳定状态。 现在,我们可以继续收集生产数据。
2.4 Production
恭喜你! 经过所有的辛苦工作,您终于可以轻松地完成工作了。
在此阶段,您基本上使用与最终平衡阶段相同的协议,并继续运行模拟,直到您满意或用完计算机时间为止。
再次,输入文件是:
Constant pressure constant temperature production run
&cntrl
nstlim=500000, dt=0.002, ntx=5, irest=1, ntpr=500, ntwr=5000, ntwx=5000,
temp0=300.0, ntt=1, tautp=2.0,
ntb=2, ntp=1,
ntc=2, ntf=2,
nrespa=1,
&end
请注意,我已将时间步数增加到500,000步。 您需要在此处做出的一项决定是要运行多长时间? 如今,发布级(publication)的工作通常需要纳秒级的模拟。 如果您打算收集几纳秒的生产数据,则最好自行调整自己的速度,并避免一次运行整个模拟的冲动。 “提交和忘记”太容易了。 密切关注模拟的进度似乎可能需要做很多工作,但最终,它确实可以避免浪费大量时间和不必要的痛苦。
2.5 Analysis
正如我之前提到的,对分子动力学轨迹的分析是一项高度定制的工作。 预期所有可能的分析方法并以收缩包装方式实施它们是不切实际的(尽管我不得不承认这听起来确实是一个吸引人的想法)。 在上一节中,我们已经看到了如何使用ptraj处理轨迹文件并计算RMSD。
2.5.1 通过追溯RMSD分析确定初始relaxation时间
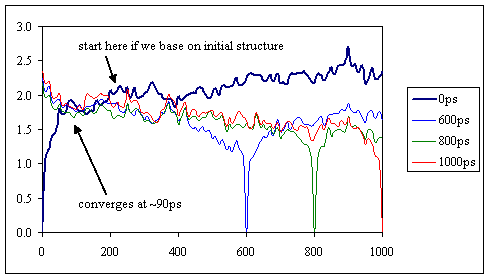
在本节中,我们将重新讨论平衡问题。最初,我们进行了有根据的猜测,并将系统平衡为100ps。然后,我们检查了势能和均方根值,并确定轨迹是稳定的,可以开始收集数据了。在论文JJ. Chem. Phys. Vol 109(23), 10115, Stella et al.指出使用从初始结构开始的均方根偏差确定初始弛豫时间的常见做法经常高估了真实值,并浪费了宝贵的仿真数据。他们提出了一种更好的方法来确定轨迹的多少真正属于初始松弛阶段,因此,提供了一种方法来恢复最初可能丢弃的某些数据。他们的方法包括使用许多后期结构作为参考来计算rmsd值,然后将它们与使用初始结构作为参考而计算出的rmsd值进行比较。通过及时跟踪曲线,我们可以更好地确定轨迹达到静止状态的点。我们将把这种分析应用于我们收集到的1-ns轨迹,包括前100 ps的恒定压力-恒定温度平衡轨迹。我已经将轨迹文件连接到一个单独的crd文件中,并在这里为您重新成像。为了减小文件大小,我还去除了轨迹中的水和抗衡离子。如果您要自己执行此操作,请不要忘记使用tleap为精简后的轨迹生成新的拓扑文件。您可以通过3个简单的步骤完成此操作:
1.采用初始拓扑并重新启动文件,并从中生成一个pdb文件:
$ ambpdb -p wt1mg.parm7 < wt1mg.rst > wt1mg_water.pdb
2.编辑wt1mg_water.pdb文件并删除所有水分子和Cl-离子。
3.使用tleap,加载此缩小的pdb文件并保存新的拓扑文件
$ tleap
> mol = loadpdb wt1mg_water.pdb
> saveAmberParm mol wt1mg_dry.parm7 wt1mg_dry.rst
> quit
用于清理轨迹的ptraj输入文件为:
trajin wt1mg_eq.crd
trajin wt1mg_md.crd
center :1-154
image center familiar
strip :WAT
strip :Cl-
rms first :3-152@CA
trajout wt1mg_dry.crd
go
您也可以从“下载”页面下载这些文件。
这是我用来完成此工作的快速而肮脏的Shell脚本:
#!/bin/csh
### take snapshots at 100ps generate reference files
cat << EOF > ptraj.in
trajin wt1mg_dry.crd 1 100 10
trajout wt1mg_ref restart
go
EOF
ptraj wt1mg_dry.parm7 ptraj.in
### calculate the rmsd for each reference structure
foreach ref (`ls wt1mg_ref*`)
set ofile=$ref.rms
cat << EOF > rms.ptraj
trajin wt1mg_dry.crd 1 100
reference $ref
rms reference out $ofile :1-154
go
EOF
ptraj wt1mg_dry.parm7 rms.ptraj
end
### clean up the files and merge them into a single file
cp wt1mg_ref.2.rms rms1
set i=10
while ($i <= 100)
cut -b12-20 wt1mg_ref.$i.rms > tmp
paste rms1 tmp > rms
cp rms rms1
@ i=$i + 10
end
rm *.rms rms1 tmp
mv rms relax.rms
运行上面的脚本后,您应该获得一个名为“ relax.rms”的输出文件。 看下面的图,得出自己的结论;-)
2.5.2 B因子计算
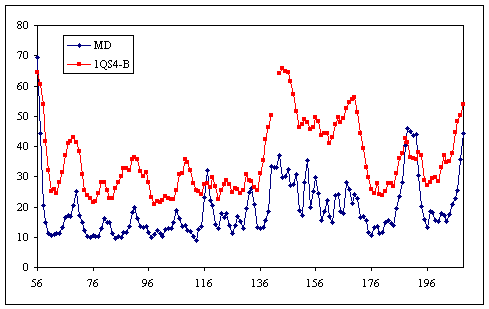
另一个常见的分析是从MD轨迹计算B因子,并将其与晶体学B因子进行比较。 用ptraj真的很容易做到
ptraj输入文件是:
trajin wt1mg_dry.crd
rms first out wt1mg_ca.b :1-154@CA
atomicfluct out wt1mg.bfactor @CA byatom bfactor
go
这将为您提供每个C-alpha原子的B因子。 我将结果与取自pdb文件1QS4链B的实验性B因子作图(这是我们用作初始模型的结构)。 您可能已经注意到,MD模拟中的B因子比晶体学B因子低得多,除了一些关键残基。 鉴于1-ns实际上并不能提供与晶体结构相比足够的采样,因此这是可以预期的。
2.5.3 可视化
有几个免费程序可以播放AMBER格式轨迹(PyMol,VMD等)。 我发现VMD在回放AMBER轨迹作为电影方面做得非常好,并且提供了许多有用的工具来交互处理数据。 我已经使用VMD在此处生成1-ns轨迹的短片供您欣赏。
您可以从MD模拟中获得什么见解? 就像在挖金一样,它们全都藏在那里,等待您发现。
2.6 下载数据地址
https://ambermd.org/tutorials/advanced/tutorial8/download.htm
参考资料
