【4.3.1】HLA分型工具--OptiType
OptiType只能分型HLA-A, HLA-B, HLA-C三种类型
- 文章2014年发表:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4441069/
- https://github.com/FRED-2/OptiType
截止2022年2月看到github上的版本:2020年9月更新到了v1.3.5
一、HLA科普
HLA(human leukocyte antigen ,⼈类⽩细胞抗原)是⼈类的主要组织相容性复合体(MHC)的表达产物。通常也 称为⼈类主要组织相容性复合物 (MHC),在染⾊体 6p21.3 上包含⼀个 4 Mb 的区域,其中包含 200 多个基因。
HLA根据不同的基因位点分为三⼤类型:I型,II型和III型。
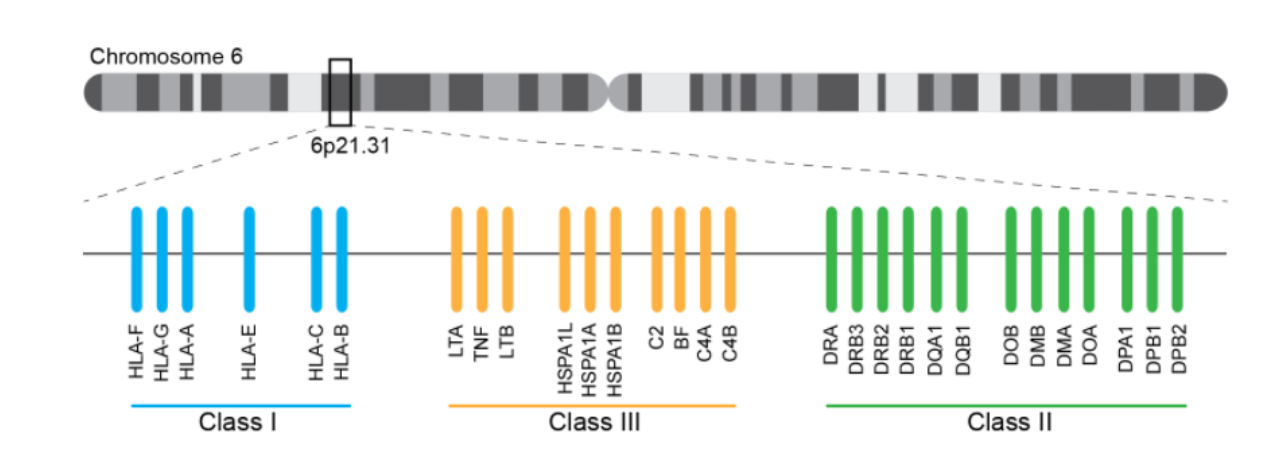
I 类分子存在于大多数细胞类型中,并存在从癌细胞、被感染细胞(病毒)的细胞内环境中衍生出来的内源性多肽。I 类分子中的结合肽呈递给 CD8+T 细胞,CD8+T 细胞会杀死受感染/受损的细胞。II 类分子主要存在于抗原呈递细胞上,并将抗原呈递给 CD4+ T 辅助细胞。III 类包含细胞因子基因和补体系统成分,它们在免疫反应中也发挥重要作用。
HLA-I型基因,包括了HLA-A,HLA-B,HLA-C等经典的抗原基因,还有一些假基因;HLA-II 型基因,其编码产物都是双链蛋白质,包括DR,DP,DQ等基因,HLA-III型基因,包含了C2,C4等补体基因,还包括其他一些基因。
HLA 复合体是整个人类基因组中多态性最强的区域,最新的统计信息如下:
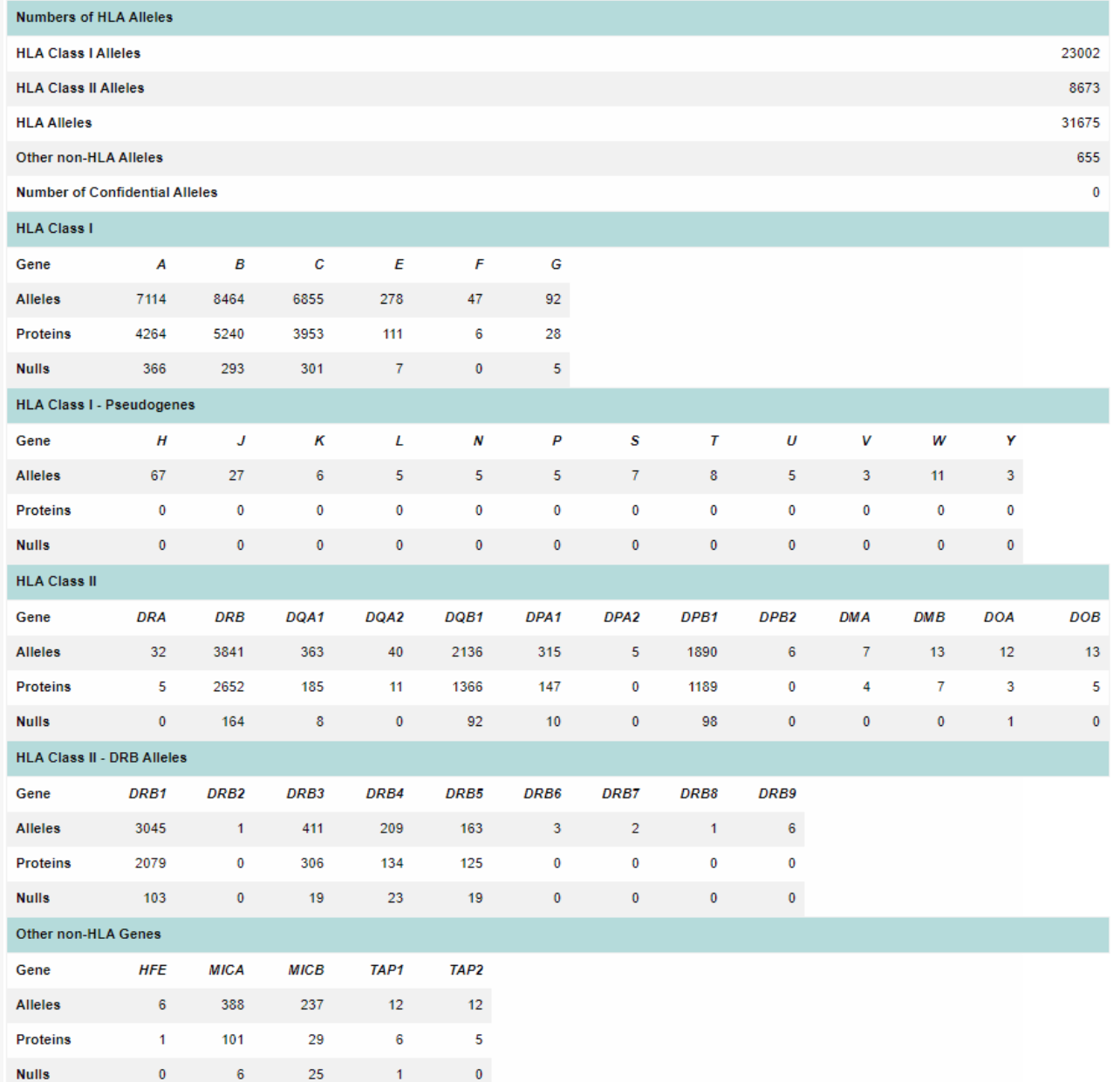
数据来自:https://www.ebi.ac.uk/ipd/imgt/hla/about/statistics/
HLA命名方式:

有众多基于全外显子组测序(WES)或全基因组测序(WGS)的HLA分型算法及工具,其中Optitype(基于整数线性规划的HLA基因分型算法)是运用的相对比较多的一款对HLA-I类分型工具
二、算法介绍
虽然下一代测序数据通常可用于患者,但推断HLA基因型是困难的,因为簇内的序列基本相似,基因座的变异性异常高
我们提出了OptiType,这是一种基于整数线性规划( integer linear programming)的新型HLA基因分型算法,能够从没有专门富集HLA聚类的NGS数据中产生准确的预测。我们还提供了一个由RNA、外显子组和全基因组测序数据组成的综合基准数据集。OptiType显著优于先前发表的硅方法,总体准确率为97%,使其能够在广泛的应用中使用
迄今已知超过7300个不同的HLA-I和2200个HLA-II等位基因[IMGT/HLA Release 3.14.02013年7月(Robinson等人,2013)]
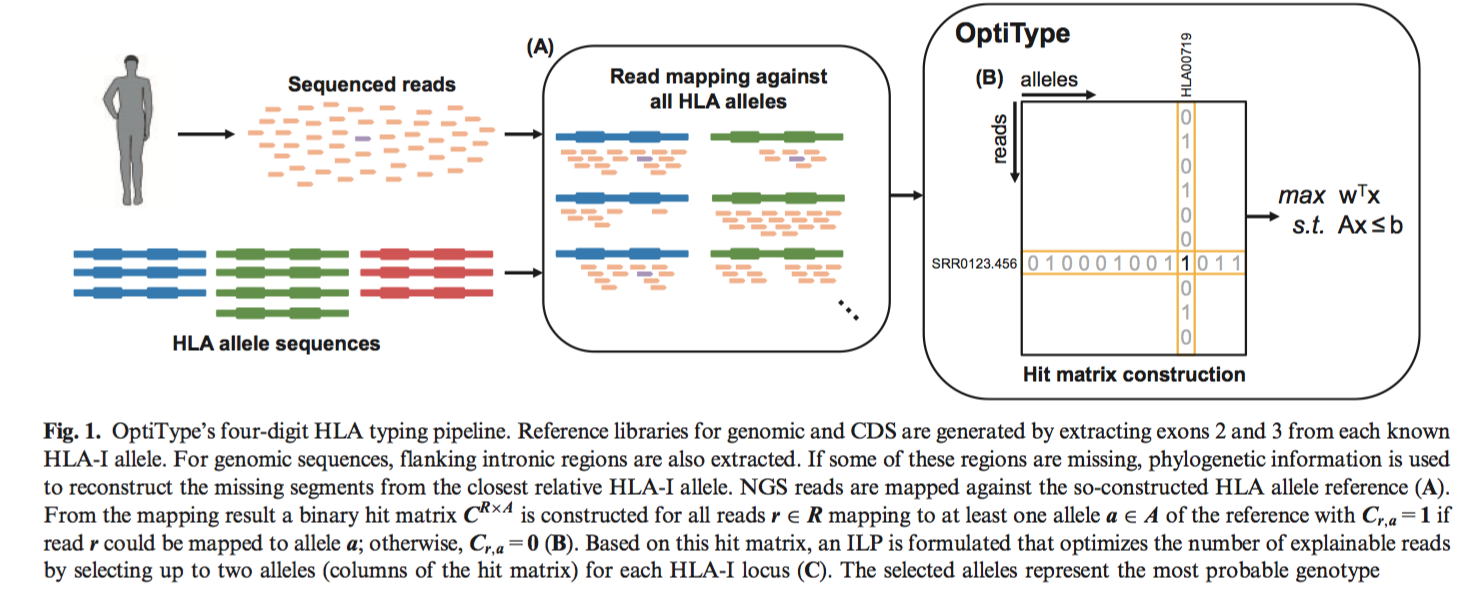
我们开发了一种名为OptiType的新方法,它同时考虑所有主要和次要的hla-1位点。OptiType工作的前提是,正确的基因型比任何其他基因型都能解释更多reads的来源,如果一个等位基因与它匹配,并且没有比任何其他等位基因更多的错配,那么这个等位基因就被称为解释了一个读取。 因此,该方法找到了一个等位基因组合,从而最大限度地增加了它们解释的读取次数。该方法包括三个关键步骤(图1)。
- 首先,根据精心构建的HLA等位基因参考(图1A)绘制读取序列。由于所有等位基因只有外显子2和3子序列可用,因此在读取映射时考虑了这些区域,因此没有等位基因因序列信息不完整而处于不利地位。此外,对于外显子组和基因组测序数据,我们包括了侧翼内含子区域,并开发了一种基于系统发育信息的方法来计算缺失的序列数据。
- 其次,从最初的读取映射结果中,生成一个二值矩阵,表示特定的读取可以与哪些等位基因对齐,错配次数最少(图1B)。
- 最后,基于这个矩阵,集合覆盖问题的一个特例(Karp, 1972)被表述为一个整数线性规划(ILP),它同时为每个基因座选择最多两个等位基因,最大限度地利用预测的基因型来解释映射的读段数量(图1C)。优化时除了考虑主要的hla - 1等位基因A、B和C外,还考虑了次要等位基因G、H和J,因为这些次要等位基因的长子序列与主要等位基因具有很高的相似性,偶尔会造成模糊的读比对
三、安装
mkdir OptiType
cd OptiType
tar -zxvf OptiType-1.3.5.tar.gz
四、使用
可以分析dna数据(WES,WGS),也可以分析RNA数据(RNAseq),用–dna和–rna参数区分。
输入为:fastq格式:
作者建议三步走:
1,使用razers3 比对原始数据(fastq/gz)到hla reference上(注意dna和rna的不同,reference放在data目录下),得到一个fished bam
>razers3 -i 95 -m 1 -dr 0 -o fished_1.bam /path/to/OptiType/data/hla_reference_dna.fasta sample_1.fastq
2,再用samtools bam2fq 将bam转换为fastq格式
>samtools bam2fq fished_1.bam > sample_1_fished.fastq
3,再用optitype
>python /path/to/OptiTypePipeline.py -i sample_fished_1.fastq [sample_fished_2.fastq]
(--rna | --dna) [--beta BETA] [--enumerate N]
[-c CONFIG] [--verbose] --outdir /path/to/out_dir/
还要提前修改 config.ini文件哦。
测试: DNA data (paired end):
python OptiTypePipeline.py -i ./test/exome/NA11995_SRR766010_1_fished.fastq ./test/exome/NA11995_SRR766010_2_fished.fastq --dna -v -o ./test/exome/
测试:RNA data()
python OptiTypePipeline.py -i ./test/rna/CRC_81_N_1_fished.fastq ./test/rna/CRC_81_N_2_fished.fastq --rna -v -o ./test/rna/
真实数据测试:PE
razers3 -i 95 -m 1 -dr 0 -o fished_1.bam /software/OptiType-1.3.5/data/hla_reference_dna.fasta 2022112_L1_1.fq.gz
razers3 -i 95 -m 1 -dr 0 -o fished_2.bam /software/OptiType-1.3.5/data/hla_reference_dna.fasta 2022112_L1_2.fq.gz
samtools bam2fq fished_1.bam > sample_1_fished.fastq
samtools bam2fq fished_2.bam > sample_2_fished.fastq
python3 /cygene/software/biosoftware/OptiType/OptiType-1.3.5/OptiTypePipeline.py -i sample_1_fished.fastq sample_2_fished.fastq --dna -v -o ./
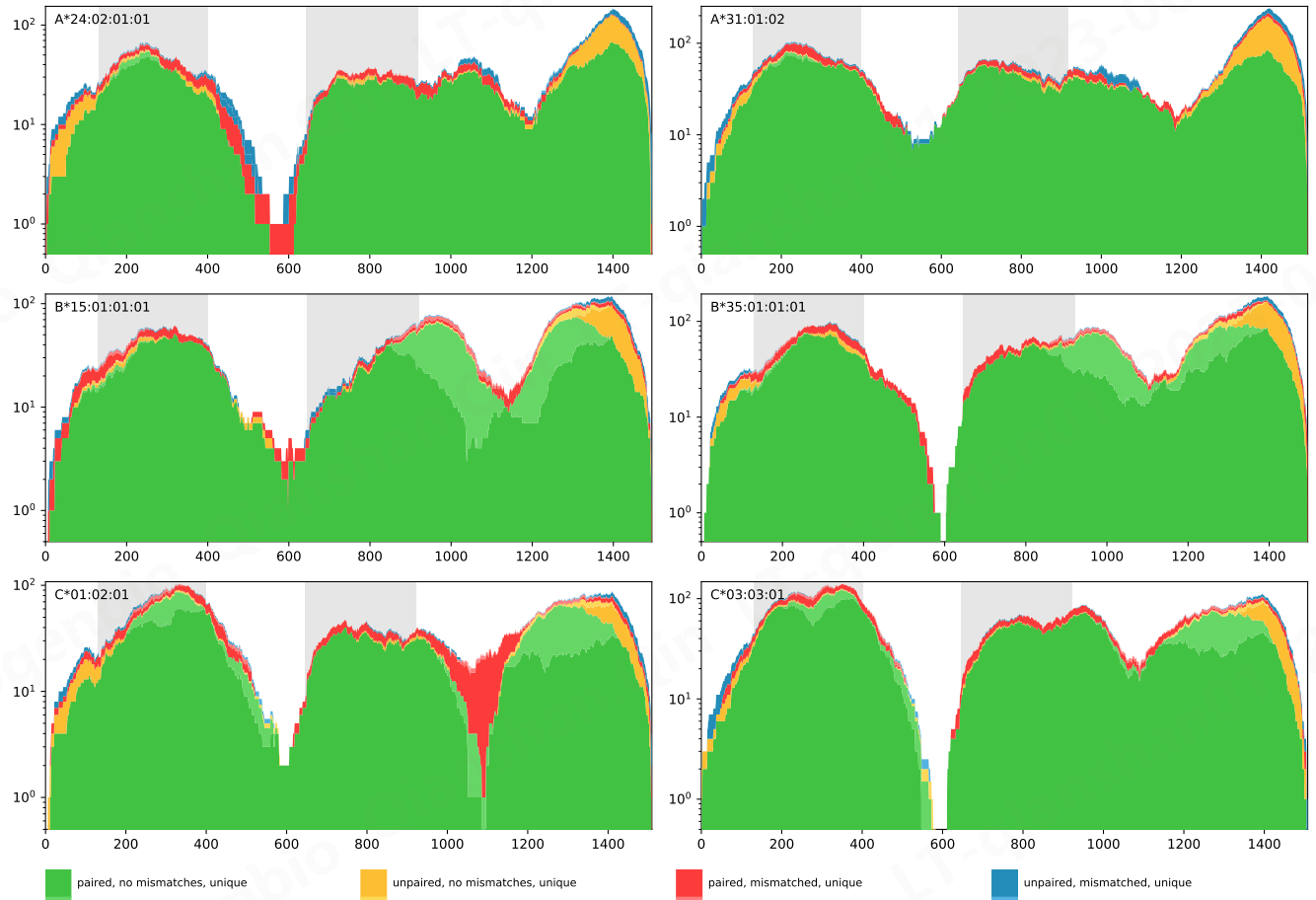
结果:一个tsv文件,和一个pdf文件如下
A1 A2 B1 B2 C1 C2 Reads Objective
0 A*24:02 A*31:01 B*15:01 B*35:01 C*01:02 C*03:03 3323.0 3203.3720000000003
参考资料
