SnapGene
一、 SnapGene中的几个View介绍
View1:Map
1.打开一个质粒图谱文件,在Topology option处选择circular。得如下界面:
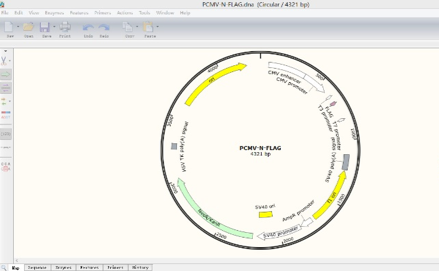
 显示质粒图谱的酶切位点。右侧箭头可显示不同厂家出售的酶。
显示质粒图谱的酶切位点。右侧箭头可显示不同厂家出售的酶。
 显示质粒图谱的开放阅读框及转录方向。点击其显示的箭头可显示该ORF的片段大小、GC%值等一些信息。
显示质粒图谱的开放阅读框及转录方向。点击其显示的箭头可显示该ORF的片段大小、GC%值等一些信息。
 显示片段名称。
显示片段名称。
△给非编码序列命名:如多克隆位点。先找到质粒图谱中的多克隆位点的第一个酶切位点和最后一个酶切位点。
点击左侧sequence ,找到两个酶切位点之间的序列。
View2:Sequence
点击Sequence,得到如下界面:
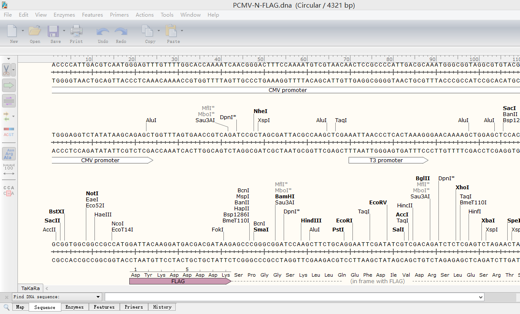
 显示编码的氨基酸序列,有缩写和全写两种。
显示编码的氨基酸序列,有缩写和全写两种。
View3:Enzymes
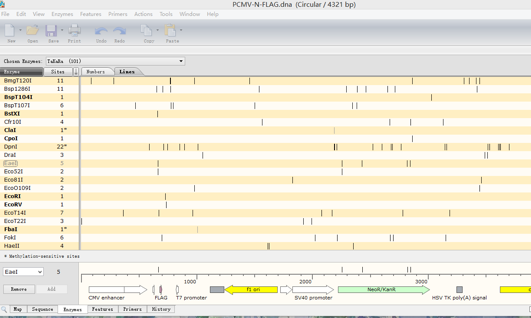
点击Enzymes,得到如下界面:
View4:Features
点击Features,得到如下界面:
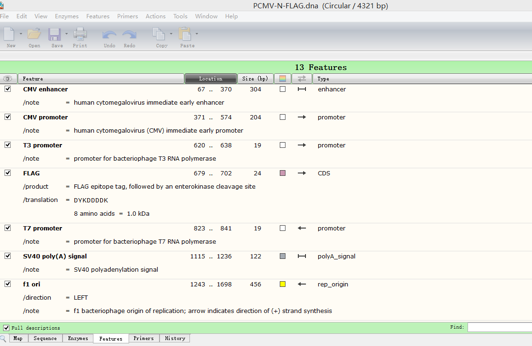
显示各个已命名片段的一些特点。
二、 对片段进行注释
1.给编码序列命名:
点击其中一个箭头,按Feature→Add Translated Feature,弹出以下窗口:
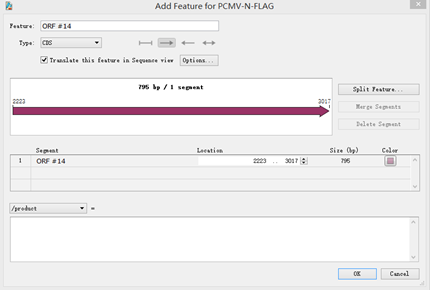
- Feature:给该片段命名。
- Type:选择该片段的类型,右侧箭头代表阅读方向。
- Color:选择颜色。
2. 给非编码序列命名:
如多克隆位点。先找到质粒图谱中的多克隆位点的第一个酶切位点和最后一个酶切位点。点击左侧sequence ,找到两个酶切位点之间的序列。
3. 给质粒图谱增加引物序列:
按Edit→Find,输入引物序列,找到质粒序列对应位置,点击Primers→Add primer,弹出该界面:
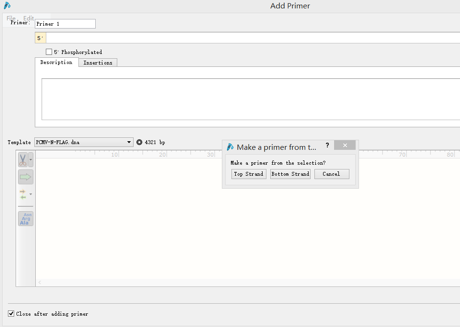
按上下游引物选择Top Strand还是Bottom Strand,在Primer处可给该引物命名,随后即可显示该引物在图谱Map中的位置。
三、 创建新DNA文件
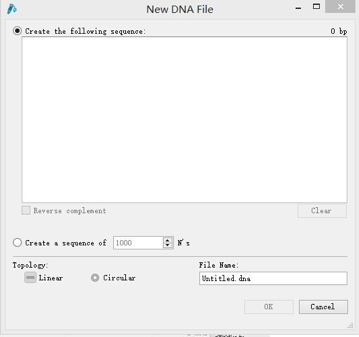
1. 打开SnapGene,点击New DNA File,弹出以下窗口:
在Create the following sequence窗口下输入DNA序列,并对该文件命名,点击OK。或是点击Import from Genebank,输入NCBI中某序列的access number,点击OK。
2.此时弹出如下窗口:
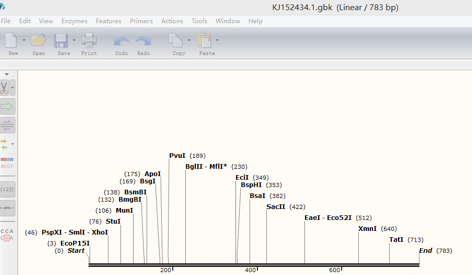
3.对该序列进行注释:点击Features→Add Feature,对该序列进行命名注释。
△创建质粒图谱文件方法相同。
四、处理序列翻译信息
1.创建一个DNA序列文件,点击 
2.显示如下箭头:
黄色箭头代表的是上面一条链编码序列,绿色箭头代表的是下面一条链编码序列。
3.点击Sequence,点击右侧箭头 ,选择All 6 Frames,可得到编码序列的所有情况,其中
,选择All 6 Frames,可得到编码序列的所有情况,其中 代表的是终止密码子。
代表的是终止密码子。
4.添加内含子:根据内含子的位置,点击Edit→Select Range,输入内含子碱基位置。点击Features→Delete Feature Segment,得到如下图:

紫色粗带为外显子,虚线为内含子部分。
五、引物、PCR和突变绘制
1.PCR引物绘制:
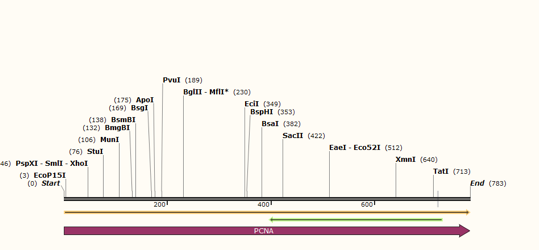
在多克隆位点处找到合适的两个酶切位点。如BamHI和XbaI,在目的基因两侧截取15-30bp序列,点击Primers→Add primers,选择top strand或bottom strand,如图:
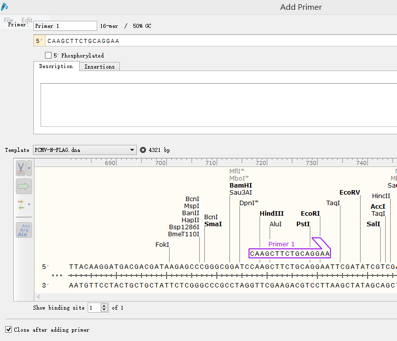
给该引物命名,然后点击Insertion,在该引物上添加之前选择好的酶切位点序列,如图选择了BamHI,点解Insert:
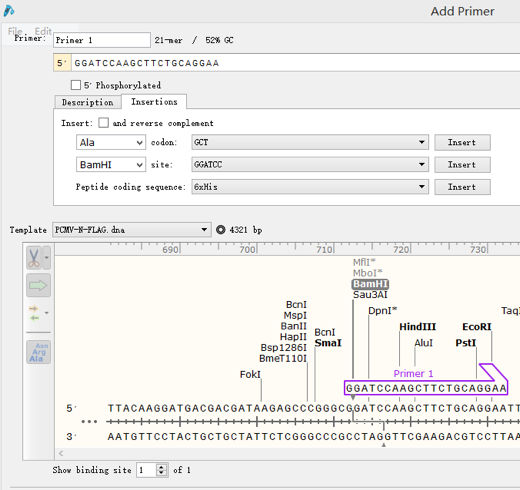
然后在该序列5’端上添加数个碱基作为保护碱基。点击完成。 △下游引物设计相同,不再赘述。
2.突变引物绘制:
在目的基因上截取一小段包含要突变位点的序列,点击Primers→Add primers,为该引物命名,在5’序列突变位点的三个碱基画黑,如图:
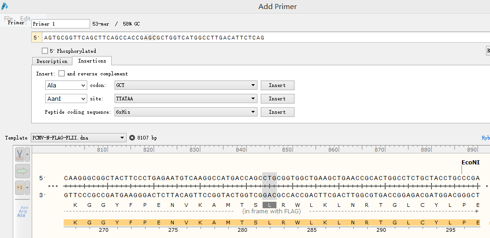
点击Insertions,选择突变成的氨基酸,点击Insert。即可获得该突变引物,点击Reverse Complement,可获得反向引物。
六、模拟标准限制性克隆
1.打开被插入片段的质粒图谱,选择合适的两个酶切位点,如HindIII和ApaI,如图:
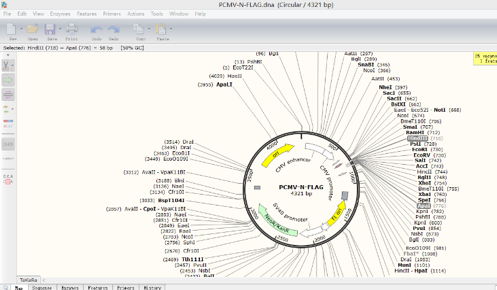
点击Actions→Insert Fragment,点击HindIII+键盘Shift键+ApaI,点击Insert,在source of fragment处选择插入的目的片段来源。点击上述同样的酶,给该重组质粒命名,点击clone。
七、 模拟融合克隆
1.打开一个需要插入片段质粒图谱,点击Actions→Insert One Fragments,弹出以下窗口:
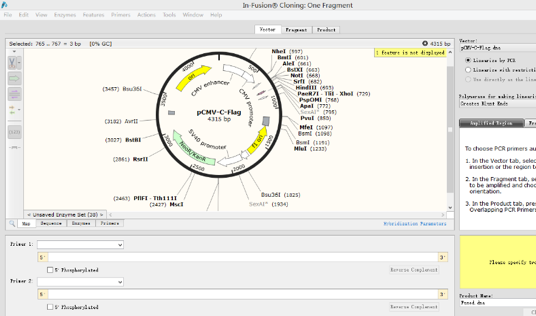
2.点击sequence,找到想要发生替换的位点。
3.点击Fragment,在Source of Fragment处选择替换片段的另外一个质粒图谱。此时弹出另外一个质粒图谱图样。
4.点击用于替换的片段,观察该片段阅读方向与被替换质粒位点的方向是否一致,如不一致,点击 更换方向。
更换方向。
5.选择后,点击Product,点击Choose Overlapping PCR Primers,此时即形成融合后的质粒图谱。
6.若想获得引物的序列,点击Primers→Export Selected Primers,选择保存。
八、 寻找序列
8.1 普通搜索
Edit - Find – Find DNA sequence
九、序列比对
9.1 参考序列比对
1.先打开参考序列(序列或者ab1文件都可以)
2.输入需要比对的序列
Tools – align to reference
9.2 多序列比对
从比对结果图中可以看到,正向测序结果(蓝色右箭头)有4个空白,即4个位点与理论序列不符;而反向测序结果(蓝色左箭头)有1个不符。接下来,详细分析这几个不匹配的位点。
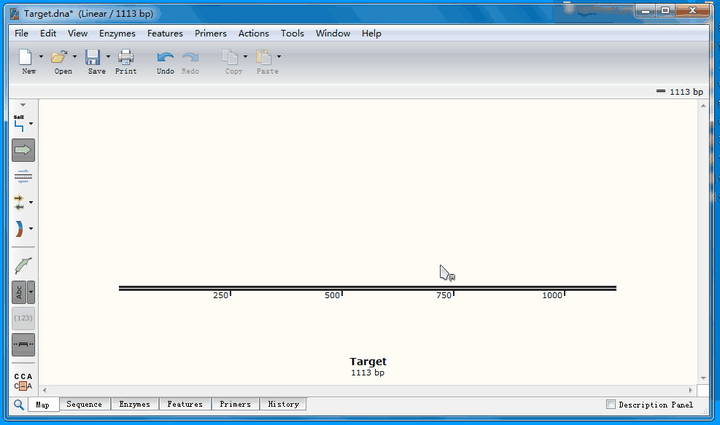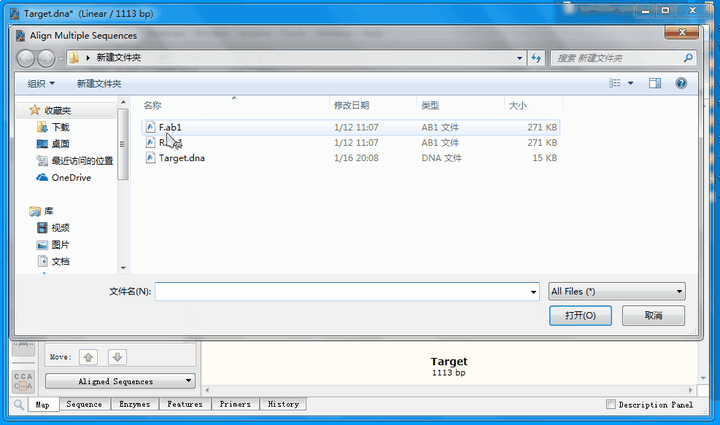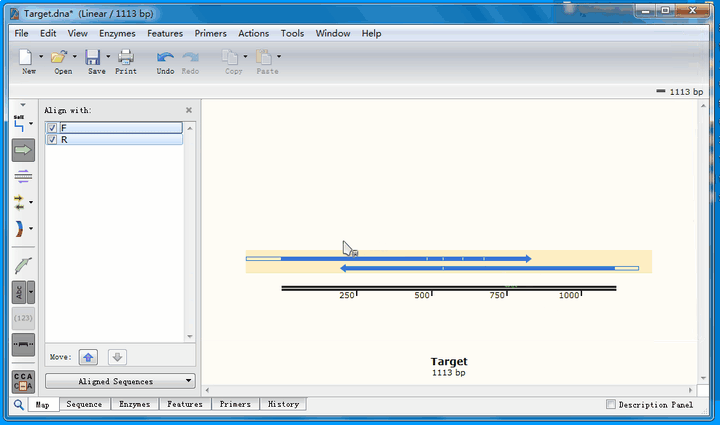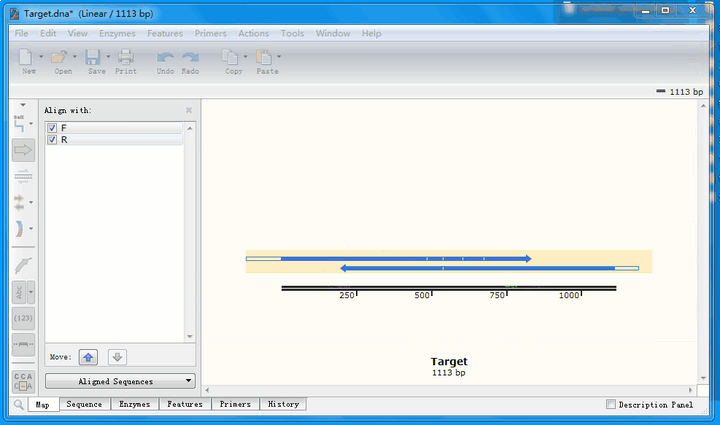
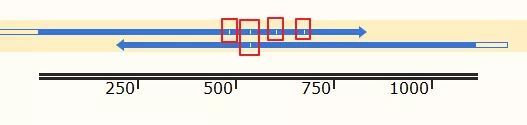
点击Sequence标签,切换到序列页面,找到不匹配的位点,这里推荐将ab1文件的峰图打开:
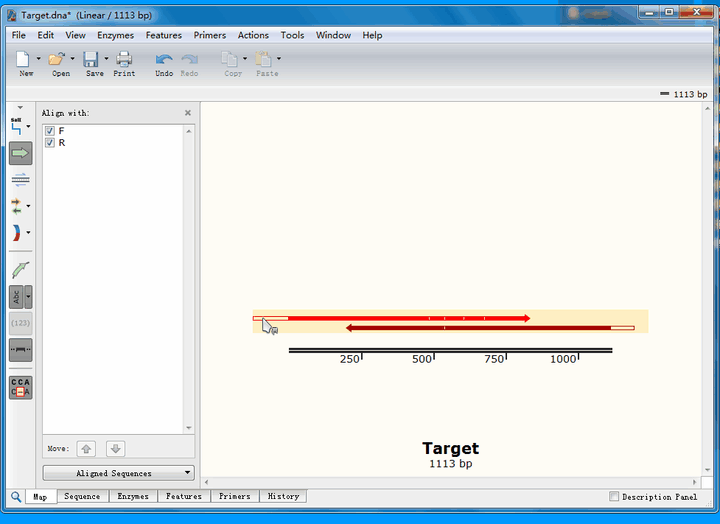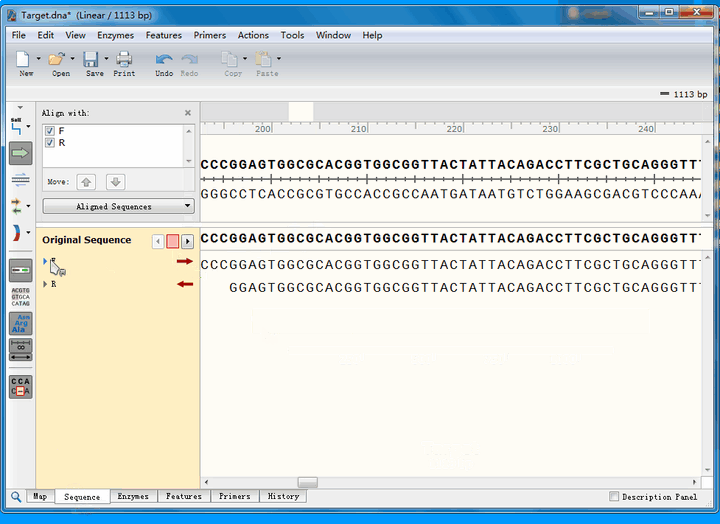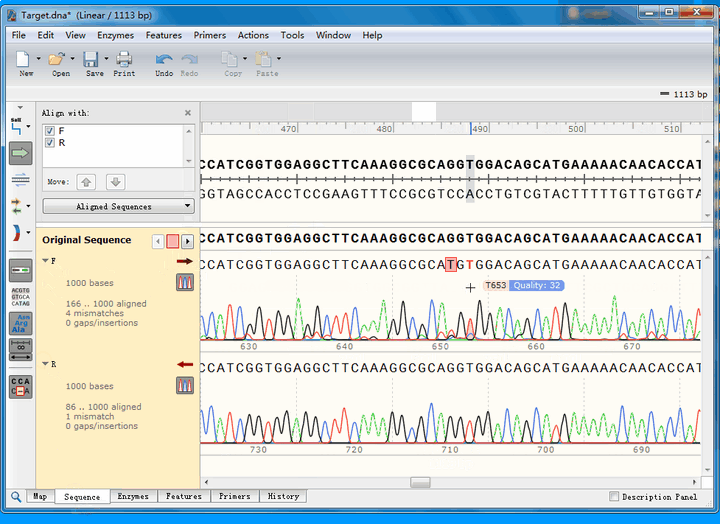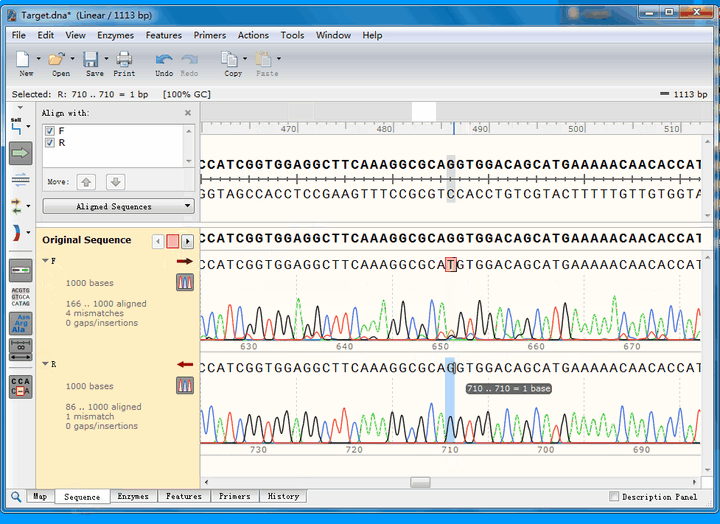
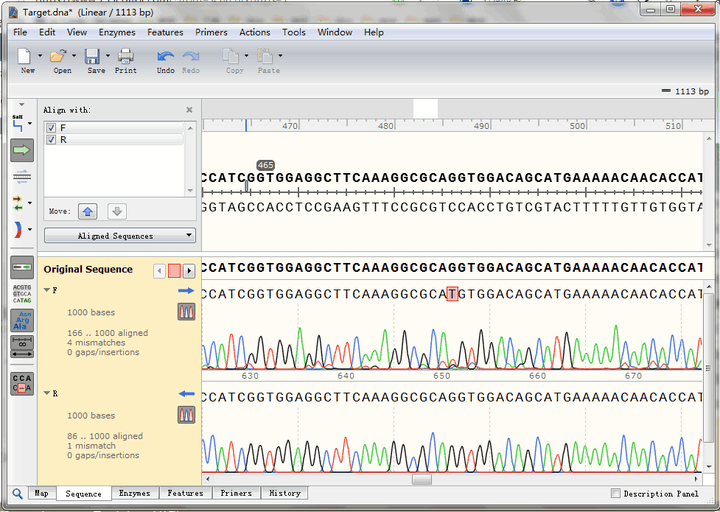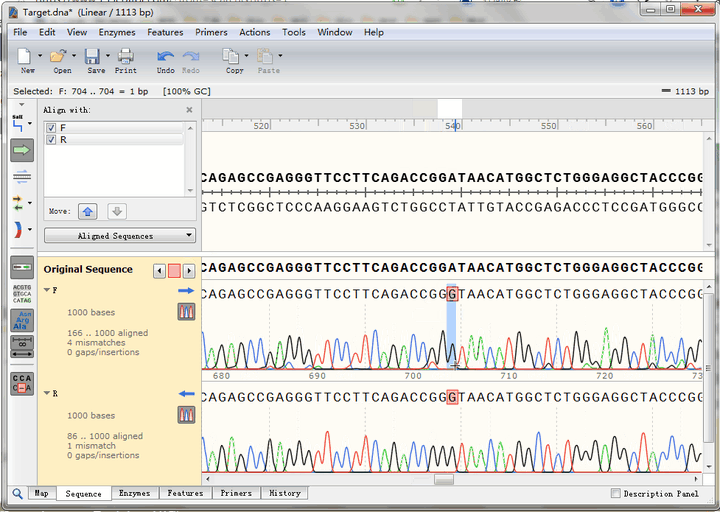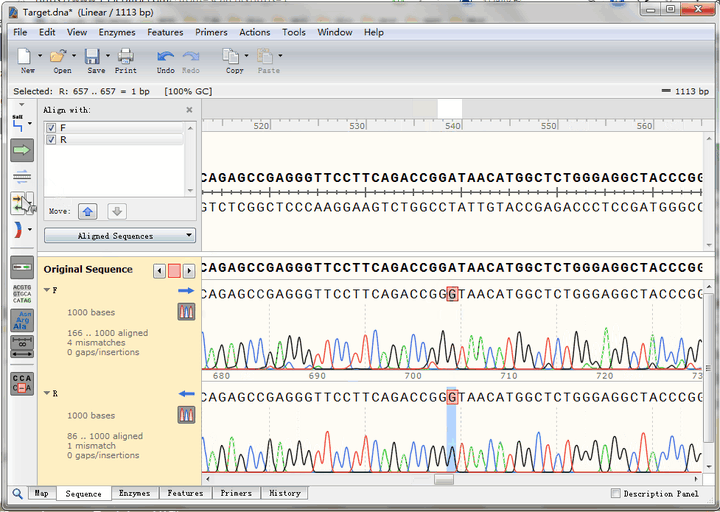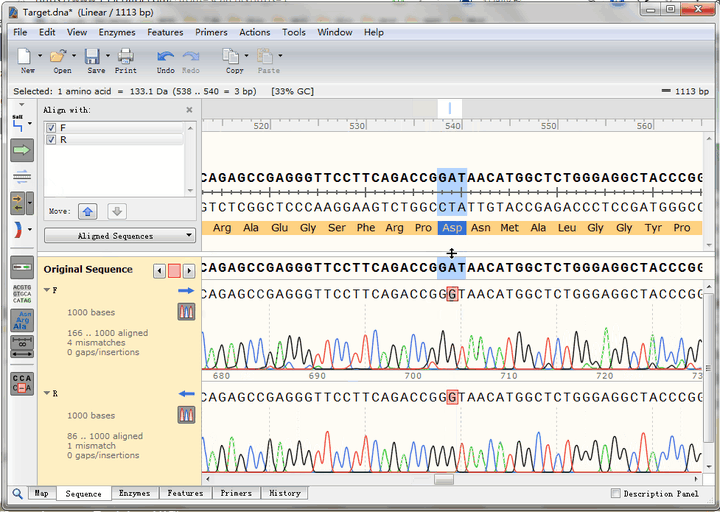
可以看到,第一个不匹配的位点,正反向测序均实现了覆盖:
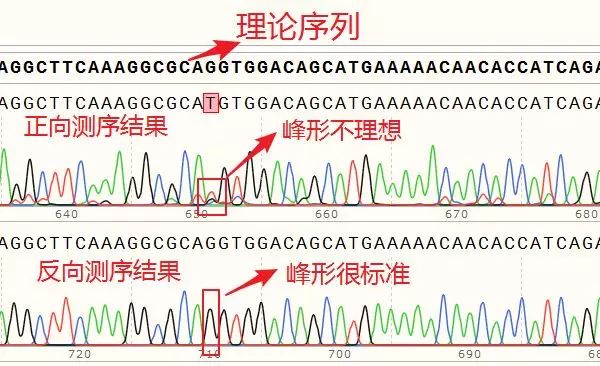
正向测序的峰高非常矮,并且出现了套峰,从而产生误读;相比之下,反向测序的峰形十分标准,因此,综合判断,该处反向测序的结果更为可信,即该处没有发生突变。
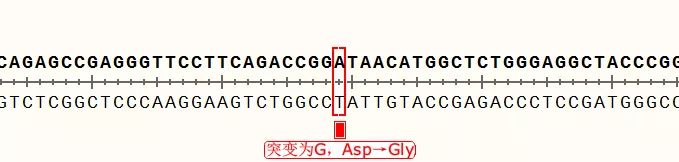
点击图中按钮,可以迅速查找到下一个不匹配碱基,可以看到,正反向测序的峰形都很标准,也就是说这里的确是实际序列与理论序列不一致,点击“预测编码蛋白”按钮,还可以看到该突变位点在CDS编码蛋白氨基酸序列中的位置,即GAT变为GGT,编码氨基酸由天冬氨酸变成了甘氨酸
可以在这里添加Feature,进行记录:
讨论
参考资料
